oggmap: Step 4 - TEI calculation
This notebook will demonstrate how to to add a transcriptome evolutionary index (short: TEI) to scRNA data.
Notebook file
Notebook file can be obtained here:
https://raw.githubusercontent.com/kullrich/oggmap/main/docs/notebooks/add_tei.ipynb
Import libraries
[1]:
import numpy as np
import pandas as pd
import scanpy as sc
import seaborn as sns
import matplotlib.pyplot as plt
from statannot import add_stat_annotation
# increase dpi
%matplotlib inline
#plt.rcParams['figure.dpi'] = 300
#plt.rcParams['savefig.dpi'] = 300
plt.rcParams['figure.figsize'] = [6, 4.5]
#plt.rcParams['figure.figsize'] = [4.4, 3.3]
Import oggmap python package submodules
[2]:
# import submodules
from oggmap import qlin, gtf2t2g, of2orthomap, orthomap2tei, datasets, ncbitax
Step 0, Step 1, Step 2 and Step 3
In order to come to Step 4, TEI calculation, one needs to have the results from Step 0, Step 1, Step 2 and Step 3.
The query species in this part is: Danio rerio (zebrafish).
Please have a look at the documentation of Step 0 - run OrthoFinder to get to know what information and files are mandatory to extract gene age classes from OrthoFinder results.
In Step 1 - get taxonomic information you have already been introduced how to extract query lineage information with oggmap and the qlin.get_qlin() function.
In Step 2 - gene age class assignment you have already been introduced how to extract an orthomap (gene age class) from OrthoFinder results with oggmap and the of2orthomap.get_orthomap() function or how to import pre-calculated orthomaps with the orthomap2tei.read_orthomap() function.
In Step 3 - map gene/transcript IDs you have already been introduced how to extract gene IDs from GTF file with orthoamp and the gtf2t2g.parse_gtf() function. You have also been introduced how to use the orthomap2tei.geneset_overlap() function to check the overlap between the gene IDs and have learned how to use the orthomap2tei.replace_by() function to e.g. reduce isoform gene IDs to gene IDs.
Step 0 - run OrthoFinder
For this documentation part all mandatory OrthoFinder (Emms and Kelly, 2019) results have been pre-calculated.
Please have a look at the documentation of Step 0 - run OrthoFinder to get further insides.
The results are available here:
https://doi.org/10.5281/zenodo.7242264
or can be accessed with the dataset submodule of oggmap
datasets.ensembl113_last(datapath='data') (download folder set to 'data').
Step 1 - get taxonomic information
Please have a look at the documentation of Step 1 - get taxonomic information to get further insides.
[3]:
# get query species taxonomic lineage information
query_lineage = qlin.get_qlin(q='Danio rerio', dbname='data/taxadb.sqlite')
query name: Danio rerio
query taxID: 7955
query kingdom: Eukaryota
query lineage names:
['root(1)', 'cellular organisms(131567)', 'Eukaryota(2759)', 'Opisthokonta(33154)', 'Metazoa(33208)', 'Eumetazoa(6072)', 'Bilateria(33213)', 'Deuterostomia(33511)', 'Chordata(7711)', 'Craniata(89593)', 'Vertebrata(7742)', 'Gnathostomata(7776)', 'Teleostomi(117570)', 'Euteleostomi(117571)', 'Actinopterygii(7898)', 'Actinopteri(186623)', 'Neopterygii(41665)', 'Teleostei(32443)', 'Osteoglossocephalai(1489341)', 'Clupeocephala(186625)', 'Otomorpha(186634)', 'Ostariophysi(32519)', 'Otophysi(186626)', 'Cypriniphysae(186627)', 'Cypriniformes(7952)', 'Cyprinoidei(30727)', 'Danionidae(2743709)', 'Danioninae(2743711)', 'Danio(7954)', 'Danio rerio(7955)']
query lineage:
[1, 131567, 2759, 33154, 33208, 6072, 33213, 33511, 7711, 89593, 7742, 7776, 117570, 117571, 7898, 186623, 41665, 32443, 1489341, 186625, 186634, 32519, 186626, 186627, 7952, 30727, 2743709, 2743711, 7954, 7955]
Step 2 - gene age class assignment
Here, oggmap use the query species information and OrthoFinder results to extract the oldest common tree node per orthogroup along a species tree and to assign this node as the gene age to the corresponding genes.
Please have a look at the documentation of Step 2 - gene age class assignment to get further insides.
Step 3 - map gene/transcript IDs
To be able to link gene ages assignments from an orthomap and gene or transcript of scRNA dataset, one needs to check the overlap of the annotated gene names. With the gtf2t2g submodule of oggmap and the gtf2t2g.parse_gtf() function, one can extract gene and transcript names from a given gene feature file (GTF).
Please have a look at the documentation of Step 3 - map gene/transcript IDs to get further insides.
Here, the pre-calculated orthomap from Danio rerio (zebrafish) obtained via Step 0, Step 1, Step 2 and Step 3 is loaded as follows:
[4]:
datasets.zebrafish_ensembl113_orthomap(datapath='data')
100% [..........................................................................] 1901053 / 1901053
[4]:
'data/zebrafish_ensembl_113_orthomap.tsv'
[5]:
# get query species orthomap
# download zebrafish orthomap results here: https://doi.org/10.5281/zenodo.7242263
# or download with datasets.zebrafish_ensembl_113_orthomap(datapath='data')
query_orthomap = orthomap2tei.read_orthomap('data/zebrafish_ensembl_113_orthomap.tsv')
query_orthomap
[5]:
| seqID | Orthogroup | PSnum | PStaxID | PSname | PScontinuity | geneID | |
|---|---|---|---|---|---|---|---|
| 0 | ENSDART00000013359.10 | OG0000000 | 10 | 7742 | Vertebrata | 0.909091 | ENSDARG00000013014 |
| 1 | ENSDART00000014270.5 | OG0000000 | 10 | 7742 | Vertebrata | 0.909091 | ENSDARG00000094080 |
| 2 | ENSDART00000099395.5 | OG0000000 | 10 | 7742 | Vertebrata | 0.909091 | ENSDARG00000068659 |
| 3 | ENSDART00000145885.2 | OG0000000 | 10 | 7742 | Vertebrata | 0.909091 | ENSDARG00000094125 |
| 4 | ENSDART00000159675.3 | OG0000000 | 10 | 7742 | Vertebrata | 0.909091 | ENSDARG00000101806 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 24956 | ENSDART00000191935.1 | OG0025717 | 25 | 30727 | Cyprinoidei | 1.000000 | ENSDARG00000114540 |
| 24957 | ENSDART00000171005.2 | OG0025718 | 22 | 186626 | Otophysi | 0.666667 | ENSDARG00000102218 |
| 24958 | ENSDART00000143229.2 | OG0025719 | 29 | 7955 | Danio rerio | 1.000000 | ENSDARG00000069978 |
| 24959 | ENSDART00000143837.3 | OG0025719 | 29 | 7955 | Danio rerio | 1.000000 | ENSDARG00000078193 |
| 24960 | ENSDART00000143384.2 | OG0025720 | 22 | 186626 | Otophysi | 0.666667 | ENSDARG00000092452 |
24961 rows × 7 columns
Import now, the scRNA dataset of the query species
Here, data is used, like in the publication (Farrell et al., 2018; Wagner et al., 2018; Qiu et al., 2022).
scRNA data was downloaded from http://tome.gs.washington.edu/ as R rds files, combined into a single Seurat object and converted into loom and AnnData (h5ad) files to be able to analyse with e.g. python scanpy or oggmap package and is available here:
https://doi.org/10.5281/zenodo.7243602
or can be accessed with the dataset submodule of oggmap:
datasets.qiu22_zebrafish(datapath='data') (download folder set to 'data').
[6]:
# load scRNA data
# download zebrafish scRNA data here: https://doi.org/10.5281/zenodo.7243602
# or download with datasets.qui22_zebrafish(datapath='data')
#zebrafish_data = datasets.qiu22_zebrafish(datapath='data')
zebrafish_data = sc.read_h5ad('data/zebrafish_data.h5ad')
#subset of the zebrafish_data
zebrafish_data_subset = zebrafish_data[:1000].copy()
Step 4 - Get TEI values and add them to scRNA dataset
Since now the gene names correspond to each other in the orthomap and the scRNA adata object, one can calculate the transcriptome evolutionary index (TEI) and add them to the scRNA dataset (adata object).
The TEI measure represents the weighted arithmetic mean (expression levels as weights for the phylostratum value) over all evolutionary age categories denoted as phylostra.
\(TEI_s = \sum (e_{is} * ps_i) / \sum e_{is}\)
, where \(TEI_s\) denotes the TEI value in developmental stage \({s, e_{is}}\) denotes the gene expression level of gene \({i}\) in stage \({s}\), and \({ps_i}\) denotes the corresponding phylostratum of gene \({i, i = 1,...,N}\) and \({N = total\ number\ of\ genes}\).
Note: If e.g. two different isoforms would fall into two different gene age classes, their gene ages might differ based on the oldest ortholog found in their corresponding orthologous groups. However, both isoforms share the same gene name and their gene ages would clash. In this case one can decide either to use the keep='min' or keep='max' gene age to be kept by the get_tei function, which defaults to keep in this cases the keep='min' or in other words the ‘older’ gene age.
To be able to re-use the original count data, they are added as a new layer to the adata object. This is useful because later on the count data can be used to extract either the relative expression per gene age class or re-calculate other metrics.
This can be done either on un-normalized counts, on normalized and log-transformed data.
[7]:
zebrafish_data.layers['counts'] = zebrafish_data.X
add TEI to adata object
Using the submodule orthomap2tei from oggmap and the orthomap2tei.get_tei() function, transcriptome evolutionary index (TEI) values are calculated and directyl added to the existing adata object (add_obs=True).
There are other options to e.g. not start from the adata.X counts but from another layer from the adata object, the default is to use the adata.X (layer=None). By default, the values are pre-processed by the normalize_total option and the log1p option.
Note: Please have a look at Liu & Robinson-Rechavi, 2018 to get more insides how expression transformation can influence TEI calculation.
If add_obs=True the resulting TEI values are added to the existing adata object as a new observation with the name set with the obs_name option.
If add_var=True the gene age values are added to the existing adata object as a new variable with the name set with the var_name option.
Note: Genes not assigned to any gene class will get a missing assignment.
If one wants to calculate bootstrap TEI values per cell, the boot option can be set to boot=True and gene age classes will be randomly chosen prior calculating TEI values bt=10 times.
Note: If memory is a problem with large annData objects, consider processing the adata object in chunks using the chunk_size parameter.
[8]:
# add TEI values to existing adata object
orthomap2tei.get_tei(adata=zebrafish_data,
gene_id=query_orthomap['geneID'],
gene_age=query_orthomap['PSnum'],
keep='min',
layer=None,
add_var=True,
var_name='Phylostrata',
add_obs=True,
obs_name='tei',
boot=False,
bt=10,
normalize_total=True,
log1p=True,
target_sum=1e6,
chunk_size=100000)
[8]:
| tei | |
|---|---|
| hpf3.3_ZFHIGH_WT_DS5_AAAAGTTGCCTC | 5.514485 |
| hpf3.3_ZFHIGH_WT_DS5_AAACAAGTGTAT | 5.503633 |
| hpf3.3_ZFHIGH_WT_DS5_AAACACCTCGTC | 5.488072 |
| hpf3.3_ZFHIGH_WT_DS5_AAATGAGGTTTN | 5.50889 |
| hpf3.3_ZFHIGH_WT_DS5_AACCCTCTCGAT | 5.574979 |
| ... | ... |
| hpf24_DEW057_TGACACAACAG_GCCACATC | 5.189284 |
| hpf24_DEW057_CTTACGGG_AACCTGAC | 5.399853 |
| hpf24_DEW057_TGAACATCTAT_GACGATGG | 5.154028 |
| hpf24_DEW057_TGAGGTTTCTC_CTCAGAAT | 5.050597 |
| hpf24_DEW057_ACGTGCTAG_CAAGTCAT | 5.344325 |
71203 rows × 1 columns
In the adata object, for each cell there is now a new variable called tei, which can be used in downstream analysis.
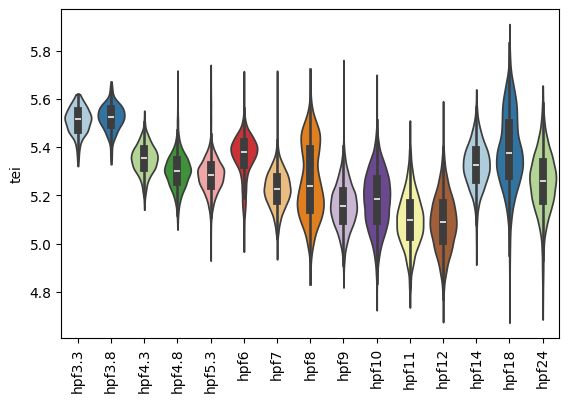
Once the gene age data has been added to the scRNA dataset, one can e.g. plot the corresponding transcriptome evolutionary index (TEI) values by any given observation pre-defined in the scRNA dataset.
Boxplot gene age class per embryo stage
[9]:
sc.pl.violin(adata=zebrafish_data,
keys=['tei'],
groupby='stage',
rotation=90,
palette='Paired',
stripplot=False,
inner='box',
hue='stage',
legend=False)
calculate bootstrap on subet of the data
[10]:
# calculate bootstrap
orthomap2tei.get_tei(adata=zebrafish_data_subset,
gene_id=query_orthomap['geneID'],
gene_age=query_orthomap['PSnum'],
keep='min',
layer=None,
add_var=True,
var_name='Phylostrata',
add_obs=True,
obs_name='tei',
boot=True,
bt=10,
normalize_total=True,
log1p=True,
target_sum=1e6,
chunk_size=100000)
|████████████████████████████████████████| 10/10 [100%] in 2.5s (4.00/s)
[10]:
( tei
hpf3.3_ZFHIGH_WT_DS5_AAAAGTTGCCTC 5.514485
hpf3.3_ZFHIGH_WT_DS5_AAACAAGTGTAT 5.503633
hpf3.3_ZFHIGH_WT_DS5_AAACACCTCGTC 5.488072
hpf3.3_ZFHIGH_WT_DS5_AAATGAGGTTTN 5.50889
hpf3.3_ZFHIGH_WT_DS5_AACCCTCTCGAT 5.574979
... ...
hpf4.3_ZFDOME_WT_DS5_CGGACCGCAGGT 5.301656
hpf4.3_ZFDOME_WT_DS5_CGGATGGCCACT 5.358101
hpf4.3_ZFDOME_WT_DS5_CGGCAATGCTTG 5.322054
hpf4.3_ZFDOME_WT_DS5_CGGCAGTGTTAC 5.351171
hpf4.3_ZFDOME_WT_DS5_CGGCCAGCGGAN 5.402065
[1000 rows x 1 columns],
boot_0 boot_1 boot_2 boot_3 \
hpf3.3_ZFHIGH_WT_DS5_AAAAGTTGCCTC 6.759911 6.647228 6.694393 6.849195
hpf3.3_ZFHIGH_WT_DS5_AAACAAGTGTAT 6.64466 6.624946 6.689043 6.722857
hpf3.3_ZFHIGH_WT_DS5_AAACACCTCGTC 6.689828 6.718553 6.771587 6.837948
hpf3.3_ZFHIGH_WT_DS5_AAATGAGGTTTN 6.78249 6.682479 6.687551 6.839673
hpf3.3_ZFHIGH_WT_DS5_AACCCTCTCGAT 6.705242 6.738627 6.700644 6.805124
... ... ... ... ...
hpf4.3_ZFDOME_WT_DS5_CGGACCGCAGGT 6.702291 6.733482 6.642275 6.853466
hpf4.3_ZFDOME_WT_DS5_CGGATGGCCACT 6.77451 6.70771 6.707631 6.725549
hpf4.3_ZFDOME_WT_DS5_CGGCAATGCTTG 6.940786 6.586875 6.724755 6.693181
hpf4.3_ZFDOME_WT_DS5_CGGCAGTGTTAC 6.753914 6.69377 6.788657 6.836734
hpf4.3_ZFDOME_WT_DS5_CGGCCAGCGGAN 6.715839 6.721093 6.690696 6.734315
boot_4 boot_5 boot_6 boot_7 \
hpf3.3_ZFHIGH_WT_DS5_AAAAGTTGCCTC 6.806379 6.684169 6.647911 6.745181
hpf3.3_ZFHIGH_WT_DS5_AAACAAGTGTAT 6.874048 6.857684 6.640579 6.835806
hpf3.3_ZFHIGH_WT_DS5_AAACACCTCGTC 6.781303 6.572003 6.632616 6.76269
hpf3.3_ZFHIGH_WT_DS5_AAATGAGGTTTN 6.712307 6.724935 6.642234 6.80512
hpf3.3_ZFHIGH_WT_DS5_AACCCTCTCGAT 6.809793 6.68517 6.684571 6.724404
... ... ... ... ...
hpf4.3_ZFDOME_WT_DS5_CGGACCGCAGGT 6.739432 6.761997 6.659034 6.740662
hpf4.3_ZFDOME_WT_DS5_CGGATGGCCACT 6.792971 6.753959 6.727792 6.888245
hpf4.3_ZFDOME_WT_DS5_CGGCAATGCTTG 6.728347 6.707649 6.727023 6.785233
hpf4.3_ZFDOME_WT_DS5_CGGCAGTGTTAC 6.904999 6.758114 6.599089 6.737372
hpf4.3_ZFDOME_WT_DS5_CGGCCAGCGGAN 6.763937 6.733328 6.556272 6.80844
boot_8 boot_9
hpf3.3_ZFHIGH_WT_DS5_AAAAGTTGCCTC 6.778171 6.647924
hpf3.3_ZFHIGH_WT_DS5_AAACAAGTGTAT 6.779376 6.633174
hpf3.3_ZFHIGH_WT_DS5_AAACACCTCGTC 6.703302 6.685941
hpf3.3_ZFHIGH_WT_DS5_AAATGAGGTTTN 6.713637 6.626618
hpf3.3_ZFHIGH_WT_DS5_AACCCTCTCGAT 6.736239 6.686084
... ... ...
hpf4.3_ZFDOME_WT_DS5_CGGACCGCAGGT 6.550693 6.772745
hpf4.3_ZFDOME_WT_DS5_CGGATGGCCACT 6.587417 6.79228
hpf4.3_ZFDOME_WT_DS5_CGGCAATGCTTG 6.635318 6.615079
hpf4.3_ZFDOME_WT_DS5_CGGCAGTGTTAC 6.828364 6.599813
hpf4.3_ZFDOME_WT_DS5_CGGCCAGCGGAN 6.743372 6.586846
[1000 rows x 10 columns])
Please have a look at the documentation of Downstream analysis to get further insides.