Case study: re-analysis of nematode (Caenorhabditis elegans) embryogenesis single-cell data
This notebook will demonstrate scRNA-seq processing with oggmap using nematode scRNA data from (Packer and Zhu al., 2019).
scRNA data were obtained from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126954, converted into Scanpy AnnData objects (Wolf et al., 2018) and are availabe here:
https://doi.org/10.5281/zenodo.7245547
or can be accessed with the dataset submodule of oggmap
datasets.packer19(datapath='data') (download folder set to 'data').
Notebook file
Notebook file can be obtained here:
https://raw.githubusercontent.com/kullrich/oggmap/main/docs/notebooks/nematode_example.ipynb
Steps
To process the scRNA data, we will do the following:
Use pre-calculated gene age classification
Get query species taxonomic lineage information
Get query species orthomap
Map OrthoFinder gene names and scRNA gene/transcript names
Get TEI values and add them to scRNA dataset
Get partial TEI values to visualize gene age class contributions
Process scRNA data and visualize TEI
Import libraries
[1]:
import numpy as np
import pandas as pd
import scanpy as sc
import seaborn as sns
import matplotlib.pyplot as plt
from statannot import add_stat_annotation
# increase dpi
%matplotlib inline
#plt.rcParams['figure.dpi'] = 300
#plt.rcParams['savefig.dpi'] = 300
#plt.rcParams['figure.figsize'] = [6, 4.5]
plt.rcParams['figure.figsize'] = [4.4, 3.3]
Import oggmap python package submodules
[2]:
# import submodules
from oggmap import qlin, gtf2t2g, of2orthomap, orthomap2tei, datasets
Step 0 - Use pre-calculated gene age classification
Orthomap was pre-calculated (Sun et al., 2021) and is available here:
https://doi.org/10.5281/zenodo.7242263
or can be accessed with the dataset submodule of oggmap
datasets.sun21_orthomap(datapath='data') (download folder set to 'data').
If you want to use your own OrthoFinder results:
oggmap can extract gene age classification from existing OrthoFinder results and link them with scRNA data.
A detailed how-to is available here:
https://oggmap.readthedocs.io/en/latest/tutorials/orthofinder.html
[3]:
# download pre-calculated orthomap into data folder
datasets.sun21_orthomap(datapath='data')
100% [........................................................] 344640 / 344640
[3]:
'data/Sun2021_Orthomap.tsv'
Step 0 - Use different pre-calculated evolutionary indices
Diversity parameter were pre-calculated (Ma et al., 2021) and is available here:
https://doi.org/10.5281/zenodo.7242263
or can be accessed with the dataset submodule of oggmap
datasets.ma21_orthomap(datapath='data') (download folder set to 'data').
[4]:
# download pre-calculated evolutionay indices into data folder
datasets.ma21_fst(datapath='data')
100% [......................................................] 1049100 / 1049100
[4]:
'data/Ma2021_Fst.tsv'
Step 1 - get query species taxonomic lineage information
Given a species name or taxonomic ID, the query species lineage information is extracted with the help of the ete3 python toolkit and the NCBI taxonomy (Huerta-Cepas et al., 2016). This information is needed alongside with the taxonomic classifications for all species used in the OrthoFinder comparison.
The oggmap submodule qlin helps to get this information for you with the qlin.get_qlin() function as follows:
[5]:
# get query species taxonomic lineage information
query_lineage = qlin.get_qlin(q='Caenorhabditis elegans')
query name: Caenorhabditis elegans
query taxID: 6239
query kingdom: Eukaryota
query lineage names:
['root(1)', 'cellular organisms(131567)', 'Eukaryota(2759)', 'Opisthokonta(33154)', 'Metazoa(33208)', 'Eumetazoa(6072)', 'Bilateria(33213)', 'Protostomia(33317)', 'Ecdysozoa(1206794)', 'Nematoda(6231)', 'Chromadorea(119089)', 'Rhabditida(6236)', 'Rhabditina(2301116)', 'Rhabditomorpha(2301119)', 'Rhabditoidea(55879)', 'Rhabditidae(6243)', 'Peloderinae(55885)', 'Caenorhabditis(6237)', 'Caenorhabditis elegans(6239)']
query lineage:
[1, 131567, 2759, 33154, 33208, 6072, 33213, 33317, 1206794, 6231, 119089, 6236, 2301116, 2301119, 55879, 6243, 55885, 6237, 6239]
Step 2 - gene age class assignment (query species orthomap)
Orthomap was pre-calculated (Sun et al., 2021; see Figure 3B) it is also available here:
https://doi.org/10.5281/zenodo.7242263
or can be accessed with the dataset submodule of oggmap
datasets.sun21_orthomap(datapath='data') (download folder set to 'data').
The pre-calculated orthomap can be imported from the orthomap2tei submodule with the orthomap2tei.read_orthomap() function as follwos:
[6]:
# get query species orthomap
# download pre-calculated orthomap here: https://doi.org/10.5281/zenodo.7242263
# or download with datasets.sun21_orthomap(datapath='data')
query_orthomap = orthomap2tei.read_orthomap(orthomapfile='data/Sun2021_Orthomap.tsv')
query_orthomap
[6]:
| GeneID | Phylostratum | |
|---|---|---|
| 0 | WBGene00000001 | 1 |
| 1 | WBGene00000002 | 1 |
| 2 | WBGene00000003 | 1 |
| 3 | WBGene00000004 | 1 |
| 4 | WBGene00000005 | 2 |
| ... | ... | ... |
| 20035 | WBGene00305132 | 9 |
| 20036 | WBGene00305157 | 1 |
| 20037 | WBGene00305158 | 13 |
| 20038 | WBGene00305159 | 11 |
| 20039 | WBGene00305173 | 0 |
20040 rows × 2 columns
Get query species evolutionary indices
[7]:
# get query species Fst values
# download pre-calculated Fst values here: https://doi.org/10.5281/zenodo.7242263
# or download with datasets.ma21_fst(datapath='data')
query_fst = pd.read_csv('data/Ma2021_Fst.tsv', delimiter='\t')
query_fst
[7]:
| WormBase_ID | Chr | Gene | TajimaD | NormalizedPi | FayWu | FST | |
|---|---|---|---|---|---|---|---|
| 0 | WBGene00000001 | I | aap-1 | -0.6957 | 0.0002 | -1.2575 | 0.8062 |
| 1 | WBGene00000002 | IV | aat-1 | -0.4724 | 0.0001 | -1.4628 | 0.8846 |
| 2 | WBGene00000003 | V | aat-2 | -1.5266 | 0.0001 | 0.0816 | 0.1691 |
| 3 | WBGene00000004 | X | aat-3 | -1.6401 | 0.0003 | -4.7685 | 0.8129 |
| 4 | WBGene00000005 | IV | aat-4 | -1.2137 | 0.0006 | -0.7617 | 0.3725 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 20217 | WBGene00271701 | X | F10D7.10 | -0.7428 | 0.0000 | -1.9308 | 0.1111 |
| 20218 | WBGene00271703 | III | ZK1010.12 | -1.3386 | 0.0008 | -3.2528 | 0.7683 |
| 20219 | WBGene00271706 | II | D2089.8 | -0.8312 | 0.0012 | 0.4489 | 0.5551 |
| 20220 | WBGene00271707 | V | ZK105.14 | -1.0748 | 0.0004 | -3.2069 | 0.6433 |
| 20221 | WBGene00271715 | III | B0244.17 | -0.6617 | 0.0010 | -1.1870 | 0.6556 |
20222 rows × 7 columns
Group evolutionary indices into bins
[8]:
# see here for additional quantile methods: https://numpy.org/doc/stable/reference/generated/numpy.nanquantile.html
orthomap2tei.get_bins(tobin_df=query_fst,
bincol='TajimaD',
q=[.1, .2, .3, .4, .5, .6, .7, .8, .9],
method='median_unbiased')
orthomap2tei.get_bins(tobin_df=query_fst,
bincol='NormalizedPi',
q=[.2, .4, .6, .8],
method='median_unbiased')
orthomap2tei.get_bins(tobin_df=query_fst,
bincol='FayWu',
q=[.1, .2, .3, .4, .5, .6, .7, .8, .9],
method='median_unbiased')
orthomap2tei.get_bins(tobin_df=query_fst,
bincol='FST',
q=[.2, .4, .6, .8],
method='median_unbiased')
[8]:
| WormBase_ID | Chr | Gene | TajimaD | NormalizedPi | FayWu | FST | TajimaD_binned | TajimaD_bins | NormalizedPi_binned | NormalizedPi_bins | FayWu_binned | FayWu_bins | FST_binned | FST_bins | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | WBGene00000001 | I | aap-1 | -0.6957 | 0.0002 | -1.2575 | 0.8062 | 8.0 | -0.84 >= x < -0.67368 | 3.0 | 0.0002 >= x < 0.0004 | 5.0 | -1.68402 >= x < -0.99135 | 5.0 | 0.6286 < x |
| 1 | WBGene00000002 | IV | aat-1 | -0.4724 | 0.0001 | -1.4628 | 0.8846 | 9.0 | -0.67368 >= x < -0.20841999999999972 | 2.0 | 0.0001 >= x < 0.0002 | 5.0 | -1.68402 >= x < -0.99135 | 5.0 | 0.6286 < x |
| 2 | WBGene00000003 | V | aat-2 | -1.5266 | 0.0001 | 0.0816 | 0.1691 | 3.0 | -1.6187 >= x < -1.4602466666666667 | 2.0 | 0.0001 >= x < 0.0002 | 8.0 | 0.03219333333333343 >= x < 0.10182000000000008 | 3.0 | 0.1347 >= x < 0.3595 |
| 3 | WBGene00000004 | X | aat-3 | -1.6401 | 0.0003 | -4.7685 | 0.8129 | 2.0 | -1.8383 >= x < -1.6187 | 3.0 | 0.0002 >= x < 0.0004 | 2.0 | -9.455993333333332 >= x < -3.9512999999999994 | 5.0 | 0.6286 < x |
| 4 | WBGene00000005 | IV | aat-4 | -1.2137 | 0.0006 | -0.7617 | 0.3725 | 5.0 | -1.3196 >= x < -1.1656 | 4.0 | 0.0004 >= x < 0.0011 | 6.0 | -0.99135 >= x < 0.0 | 4.0 | 0.3595 >= x < 0.6286 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 20217 | WBGene00271701 | X | F10D7.10 | -0.7428 | 0.0000 | -1.9308 | 0.1111 | 8.0 | -0.84 >= x < -0.67368 | 1.0 | x < 0.0001 | 4.0 | -2.277410000000001 >= x < -1.68402 | 2.0 | 0.0 >= x < 0.1347 |
| 20218 | WBGene00271703 | III | ZK1010.12 | -1.3386 | 0.0008 | -3.2528 | 0.7683 | 4.0 | -1.4602466666666667 >= x < -1.3196 | 4.0 | 0.0004 >= x < 0.0011 | 3.0 | -3.9512999999999994 >= x < -2.277410000000001 | 5.0 | 0.6286 < x |
| 20219 | WBGene00271706 | II | D2089.8 | -0.8312 | 0.0012 | 0.4489 | 0.5551 | 8.0 | -0.84 >= x < -0.67368 | 5.0 | 0.0011 < x | 10.0 | 0.2674900000000009 < x | 4.0 | 0.3595 >= x < 0.6286 |
| 20220 | WBGene00271707 | V | ZK105.14 | -1.0748 | 0.0004 | -3.2069 | 0.6433 | 6.0 | -1.1656 >= x < -1.0416 | 4.0 | 0.0004 >= x < 0.0011 | 3.0 | -3.9512999999999994 >= x < -2.277410000000001 | 5.0 | 0.6286 < x |
| 20221 | WBGene00271715 | III | B0244.17 | -0.6617 | 0.0010 | -1.1870 | 0.6556 | 9.0 | -0.67368 >= x < -0.20841999999999972 | 4.0 | 0.0004 >= x < 0.0011 | 5.0 | -1.68402 >= x < -0.99135 | 5.0 | 0.6286 < x |
20222 rows × 15 columns
Gene age assignments per query species lineage node
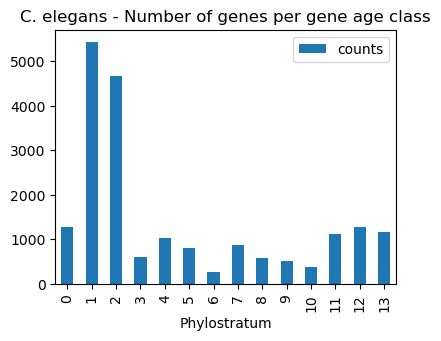
Given an orthomap, one can get an overview of the gene age assignments per query species lineage node.
The oggmap submodule of2orhomap and the of2orthomap.get_counts_per_ps() function will show the distribution of the gene age classes and can be further visualized as follows:
[9]:
# show count per taxonomic group (PStaxID)
of2orthomap.get_counts_per_ps(omap_df=query_orthomap,
psnum_col='Phylostratum',
pstaxid_col=None,
psname_col=None)
# bar plot count per taxonomic group (PSname)
ax = of2orthomap.get_counts_per_ps(omap_df=query_orthomap,
psnum_col='Phylostratum',
pstaxid_col=None,
psname_col=None).plot.bar(y='counts', x='Phylostratum')
ax.set_title('C. elegans - Number of genes per gene age class')
plt.show()
Gene assignments per query species evolutionary index
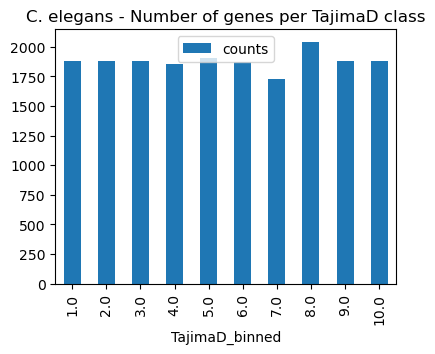
[10]:
# show count per TajimaD group (TajimaD_binned)
of2orthomap.get_counts_per_ps(omap_df=query_fst,
psnum_col='TajimaD_binned',
pstaxid_col=None,
psname_col=None)
# bar plot count per taxonomic group (PSname)
ax = of2orthomap.get_counts_per_ps(omap_df=query_fst,
psnum_col='TajimaD_binned',
pstaxid_col=None,
psname_col=None).plot.bar(y='counts', x='TajimaD_binned')
ax.set_title('C. elegans - Number of genes per TajimaD class')
plt.show()
Step 3 - map OrthoFinder gene names and scRNA gene/transcript names
To be able to link gene ages assignments from an orthomap and gene or transcript of scRNA dataset, one needs to check the overlap of the annotated gene names. With the gtf2t2g submodule of oggmap and the gtf2t2g.parse_gtf() function, one can extract gene and transcript names from a given gene feature file (GTF).
Here, pre-calculated orthomap gene names already overlap, so no GTF import is necessary (Sun et al., 2021).
Import now, the scRNA dataset of the query species
Here, data is used, like in the original publication (Packer and Zhu al., 2019).
scRNA data were downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126954 converted into Seurat object and converted into loom and AnnData (h5ad) files to be able to analyse with e.g. python scanpy or oggmap package and is availbale here:
https://doi.org/10.5281/zenodo.7245547
or can be accessed with the dataset submodule of oggmap:
datasets.packer19(datapath='data') (download folder set to 'data').
Note: A smaller scRNA data set for the same data exist and can be obtained via:
datasets.packer19_small(datapath='data') (download folder set to 'data').
[11]:
# load scRNA data
# download zebrafish scRNA data here: https://doi.org/10.5281/zenodo.7245547
# or download with datasets.packer19('data')
#celegans_data = datasets.packer19(datapath='data')
celegans_data = sc.read('data/GSE126954.h5ad')
Get an overview of observations
[12]:
celegans_data
[12]:
AnnData object with n_obs × n_vars = 89701 × 20222
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'cell', 'n.umi', 'time.point', 'batch', 'Size_Factor', 'cell.type', 'cell.subtype', 'plot.cell.type', 'raw.embryo.time', 'embryo.time', 'embryo.time.bin', 'raw.embryo.time.bin', 'lineage', 'passed_initial_QC_or_later_whitelisted'
var: 'features', 'genes'
[13]:
celegans_data.obs
[13]:
| orig.ident | nCount_RNA | nFeature_RNA | cell | n.umi | time.point | batch | Size_Factor | cell.type | cell.subtype | plot.cell.type | raw.embryo.time | embryo.time | embryo.time.bin | raw.embryo.time.bin | lineage | passed_initial_QC_or_later_whitelisted | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAACCTGAGACAATAC-300.1.1 | 0 | 1630.0 | 781 | AAACCTGAGACAATAC-300.1.1 | 1630 | 300_minutes | Waterston_300_minutes | 1.023195 | Body_wall_muscle | BWM_head_row_1 | BWM_head_row_1 | 360 | 380.0 | 330-390 | 330-390 | MSxpappp | 1 |
| AAACCTGAGGGCTCTC-300.1.1 | 0 | 2323.0 | 1116 | AAACCTGAGGGCTCTC-300.1.1 | 2319 | 300_minutes | Waterston_300_minutes | 1.458210 | NA | NA | NA | 260 | 220.0 | 210-270 | 210-270 | MSxapaap | 1 |
| AAACCTGAGTGCGTGA-300.1.1 | 0 | 3725.0 | 1322 | AAACCTGAGTGCGTGA-300.1.1 | 3719 | 300_minutes | Waterston_300_minutes | 2.338283 | NA | NA | NA | 270 | 230.0 | 210-270 | 270-330 | NA | 1 |
| AAACCTGAGTTGAGTA-300.1.1 | 0 | 4236.0 | 1747 | AAACCTGAGTTGAGTA-300.1.1 | 4251 | 300_minutes | Waterston_300_minutes | 2.659051 | Body_wall_muscle | BWM_anterior | BWM_anterior | 260 | 280.0 | 270-330 | 210-270 | Dxap | 1 |
| AAACCTGCAAGACGTG-300.1.1 | 0 | 1003.0 | 621 | AAACCTGCAAGACGTG-300.1.1 | 1003 | 300_minutes | Waterston_300_minutes | 0.629610 | Ciliated_amphid_neuron | AFD | AFD | 350 | 350.0 | 330-390 | 330-390 | ABalpppapav/ABpraaaapav | 1 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| TCTGAGACATGTCGAT-b02 | 0 | 581.0 | 361 | TCTGAGACATGTCGAT-b02 | 585 | mixed | Murray_b02 | 0.364709 | Rectal_gland | Rectal_gland | Rectal_gland | 390 | 700.0 | > 650 | 390-450 | NA | 1 |
| TCTGAGACATGTCTCC-b02 | 0 | 516.0 | 327 | TCTGAGACATGTCTCC-b02 | 510 | mixed | Murray_b02 | 0.323907 | NA | NA | NA | 510 | 470.0 | 450-510 | 510-580 | NA | 1 |
| TGGCCAGCACGAAGCA-b02 | 0 | 843.0 | 543 | TGGCCAGCACGAAGCA-b02 | 843 | mixed | Murray_b02 | 0.529174 | NA | NA | NA | 400 | 470.0 | 450-510 | 390-450 | NA | 1 |
| TGGCGCACAGGCAGTA-b02 | 0 | 634.0 | 397 | TGGCGCACAGGCAGTA-b02 | 636 | mixed | Murray_b02 | 0.397979 | NA | NA | NA | 330 | 350.0 | 330-390 | 330-390 | NA | 1 |
| TGGGCGTTCAGGCCCA-b02 | 0 | 1126.0 | 702 | TGGGCGTTCAGGCCCA-b02 | 1132 | mixed | Murray_b02 | 0.706820 | NA | NA | NA | 260 | 265.0 | 210-270 | 210-270 | NA | 1 |
89701 rows × 17 columns
[14]:
celegans_data.obs.dtypes
[14]:
orig.ident int32
nCount_RNA float64
nFeature_RNA int32
cell object
n.umi int32
time.point category
batch category
Size_Factor float64
cell.type category
cell.subtype category
plot.cell.type category
raw.embryo.time int32
embryo.time float64
embryo.time.bin category
raw.embryo.time.bin category
lineage category
passed_initial_QC_or_later_whitelisted int32
dtype: object
Prior any analysis the observations 'embryo.time.bin' and 'batch' will be converted into the 'category' type. In addition a new observation 'cell.type.per.embryo.time.bin.cat' will be created that combines sample timepoint and assigned cell type.
[15]:
# add embryo.time.bin as category
celegans_data.obs['embryo.time.bin.cat'] = celegans_data.obs['embryo.time.bin'].astype('category')
celegans_data.obs['embryo.time.bin.cat'] = celegans_data.obs['embryo.time.bin.cat'].cat.reorder_categories(['< 100',
'100-130','130-170','170-210','210-270','270-330','330-390','390-450','450-510','510-580','580-650','> 650'])
celegans_data.obs['batch.cat'] = celegans_data.obs['batch'].astype('category')
[16]:
celegans_data.obs['cell.type.per.embryo.time.bin.cat'] =\
(celegans_data.obs['cell.type'].astype('string') +\
'-' +\
celegans_data.obs['embryo.time.bin.cat'].astype('string')).astype('category')
Helper functions to match gene names
The orthomap2tei submodule contains the orthomap2tei.geneset_overlap() helper function to check for gene name overlap between the constructed orthomap from OrthoFinder results and a given scRNA dataset.
[17]:
# check overlap of orthomap <seqID> and scRNA data <var_names>
orthomap2tei.geneset_overlap(geneset1=celegans_data.var_names,
geneset2=query_orthomap['GeneID'])
[17]:
| g1_g2_overlap | g1_ratio | g2_ratio | |
|---|---|---|---|
| 0 | 19795 | 0.978884 | 0.987774 |
step 4 - Get TEI values and add them to scRNA dataset
Since now the gene names correspond to each other in the orthomap and the scRNA adata object, one can calculate the transcriptome evolutionary index (TEI) and add them to the scRNA dataset (adata object).
The TEI measure represents the weighted arithmetic mean (expression levels as weights for the phylostratum value) over all evolutionary age categories denoted as phylostra.
\({TEI_s = \sum (e_{is} * ps_i) / \sum e_{is}}\)
, where \({TEI_s}\) denotes the TEI value in developmental stage \({s, e_{is}}\) denotes the gene expression level of gene \({i}\) in stage \({s}\), and \({ps_i}\) denotes the corresponding phylostratum of gene \({i, i = 1,...,N}\) and \({N = total\ number\ of\ genes}\).
Note: If e.g. two different isoforms would fall into two different gene age classes, their gene ages might differ based on the oldest ortholog found in their corresponding orthologous groups. However, both isoforms share the same gene name and their gene ages would clash. In this case one can decide either to use the keep='min' or keep='max' gene age to be kept by the get_tei function, which defaults to keep in this cases the keep='min' or in other words the ‘older’ gene age.
To be able to re-use the original count data, they are added as a new layer to the adata object. This is useful because later on the count data can be used to extract either the relative expression per gene age class.
This can be done either on un-normalized counts, on normalized and log-transformed data.
[18]:
celegans_data.layers['counts'] = celegans_data.X
add TEI to adata object
Using the submodule orthomap2tei from oggmap and the orthomap2tei.get_tei() function, transcriptome evolutionary index (TEI) values are calculated and directyl added to the existing adata object (add_obs=True).
There are other options to e.g. not start from the adata.X counts but from another layer from the adata object, the default is to use the adata.X (layer=None). The values can be pre-processed by the normalize_total option and the log1p option.
If add_obs=True the resulting TEI values are added to the existing adata object as a new observation with the name set with the obs_name option.
If add_var=True the gene age values are added to the existing adata object as a new variable with the name set with the var_name option.
Note: Genes not assigned to any gene class will get a missing assignment.
If one wants to calculate bootstrap TEI values per cell, the boot option can be set to boot=True and gene age classes will be randomly chosen prior calculating TEI values bt=10 times.
[19]:
# add TEI values to existing adata object
orthomap2tei.get_tei(adata=celegans_data,
gene_id=query_orthomap['GeneID'],
gene_age=query_orthomap['Phylostratum'],
keep='min',
layer=None,
add_var=True,
var_name='Phylostrata',
add_obs=True,
obs_name='tei',
boot=False,
bt=10,
normalize_total=True,
log1p=True,
target_sum=1e6)
[19]:
| tei | |
|---|---|
| AAACCTGAGACAATAC-300.1.1 | 1.876533 |
| AAACCTGAGGGCTCTC-300.1.1 | 2.239562 |
| AAACCTGAGTGCGTGA-300.1.1 | 2.126684 |
| AAACCTGAGTTGAGTA-300.1.1 | 2.362362 |
| AAACCTGCAAGACGTG-300.1.1 | 2.581323 |
| ... | ... |
| TCTGAGACATGTCGAT-b02 | 1.964531 |
| TCTGAGACATGTCTCC-b02 | 2.155930 |
| TGGCCAGCACGAAGCA-b02 | 2.262958 |
| TGGCGCACAGGCAGTA-b02 | 2.565133 |
| TGGGCGTTCAGGCCCA-b02 | 2.269434 |
89701 rows × 1 columns
add TajimaD, Fst and NormalizedPi to adata object
[20]:
# add TajimaD binned values to existing adata object
orthomap2tei.get_tei(adata=celegans_data,
gene_id=query_fst['WormBase_ID'],
gene_age=query_fst['TajimaD_binned'],
keep='min',
layer=None,
add_var=True,
var_name='TajimaD_bin',
add_obs=True,
obs_name='TajimaD',
boot=False,
bt=10,
normalize_total=True,
log1p=True,
target_sum=1e6)
[20]:
| TajimaD | |
|---|---|
| AAACCTGAGACAATAC-300.1.1 | 5.769133 |
| AAACCTGAGGGCTCTC-300.1.1 | 5.898489 |
| AAACCTGAGTGCGTGA-300.1.1 | 5.736505 |
| AAACCTGAGTTGAGTA-300.1.1 | 5.795881 |
| AAACCTGCAAGACGTG-300.1.1 | 5.746057 |
| ... | ... |
| TCTGAGACATGTCGAT-b02 | 5.465485 |
| TCTGAGACATGTCTCC-b02 | 5.543912 |
| TGGCCAGCACGAAGCA-b02 | 5.524013 |
| TGGCGCACAGGCAGTA-b02 | 5.652469 |
| TGGGCGTTCAGGCCCA-b02 | 5.634361 |
89701 rows × 1 columns
[21]:
# add Fst binned values to existing adata object
orthomap2tei.get_tei(adata=celegans_data,
gene_id=query_fst['WormBase_ID'],
gene_age=query_fst['FST_binned'],
keep='min',
layer=None,
add_var=True,
var_name='FST_bin',
add_obs=True,
obs_name='Fst',
boot=False,
bt=10,
normalize_total=True,
log1p=True,
target_sum=1e6)
[21]:
| Fst | |
|---|---|
| AAACCTGAGACAATAC-300.1.1 | 2.997840 |
| AAACCTGAGGGCTCTC-300.1.1 | 3.103704 |
| AAACCTGAGTGCGTGA-300.1.1 | 3.100244 |
| AAACCTGAGTTGAGTA-300.1.1 | 3.105081 |
| AAACCTGCAAGACGTG-300.1.1 | 3.011721 |
| ... | ... |
| TCTGAGACATGTCGAT-b02 | 3.011302 |
| TCTGAGACATGTCTCC-b02 | 3.134352 |
| TGGCCAGCACGAAGCA-b02 | 3.054067 |
| TGGCGCACAGGCAGTA-b02 | 3.117862 |
| TGGGCGTTCAGGCCCA-b02 | 3.149005 |
89701 rows × 1 columns
[22]:
# add NormalizedPi binned values to existing adata object
orthomap2tei.get_tei(adata=celegans_data,
gene_id=query_fst['WormBase_ID'],
gene_age=query_fst['NormalizedPi_binned'],
keep='min',
layer=None,
add_var=True,
var_name='NormalizedPi_bin',
add_obs=True,
obs_name='NormalizedPi',
boot=False,
bt=10,
normalize_total=True,
log1p=True,
target_sum=1e6)
[22]:
| NormalizedPi | |
|---|---|
| AAACCTGAGACAATAC-300.1.1 | 2.321107 |
| AAACCTGAGGGCTCTC-300.1.1 | 2.527517 |
| AAACCTGAGTGCGTGA-300.1.1 | 2.445354 |
| AAACCTGAGTTGAGTA-300.1.1 | 2.507382 |
| AAACCTGCAAGACGTG-300.1.1 | 2.655676 |
| ... | ... |
| TCTGAGACATGTCGAT-b02 | 2.267350 |
| TCTGAGACATGTCTCC-b02 | 2.484896 |
| TGGCCAGCACGAAGCA-b02 | 2.378922 |
| TGGCGCACAGGCAGTA-b02 | 2.483049 |
| TGGGCGTTCAGGCCCA-b02 | 2.470968 |
89701 rows × 1 columns
Step 5 - downstream analysis
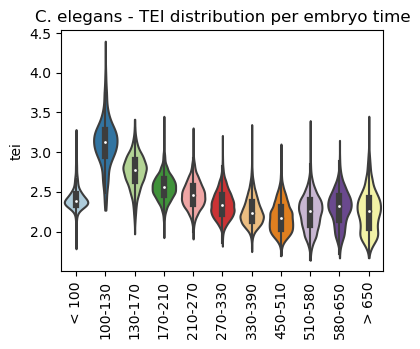
Once the gene age data has been added to the scRNA dataset, one can e.g. plot the corresponding transcriptome evolutionary index (TEI) values by any given observation pre-defined in the scRNA dataset.
Here, we plot them against the assigned sample timepoint and against assigned cell types of the nematode using the scanpy sc.pl.violin() function as follows:
Boxplot gene age class per sample timepoint
[23]:
ax = sc.pl.violin(adata=celegans_data,
keys=['tei'],
groupby='embryo.time.bin.cat',
rotation=90,
palette='Paired',
stripplot=False,
inner='box',
order=['< 100', '100-130', '130-170', '170-210',
'210-270', '270-330', '330-390', '450-510',
'510-580', '580-650', '> 650'],
show=False)
ax.set_title('C. elegans - TEI distribution per embryo time')
plt.show()
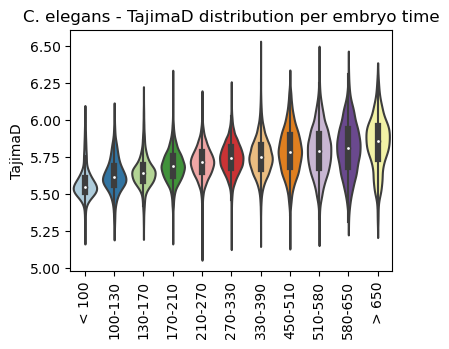
Boxplot TajimaD class per sample timepoint
[24]:
ax = sc.pl.violin(adata=celegans_data,
keys=['TajimaD'],
groupby='embryo.time.bin.cat',
rotation=90,
palette='Paired',
stripplot=False,
inner='box',
order=['< 100', '100-130', '130-170', '170-210',
'210-270', '270-330', '330-390', '450-510',
'510-580', '580-650', '> 650'],
show=False)
ax.set_title('C. elegans - TajimaD distribution per embryo time')
plt.show()
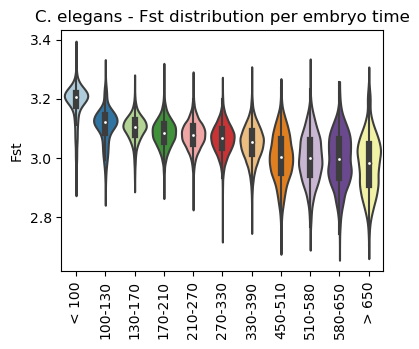
Boxplot Fst class per sample timepoint
[25]:
ax = sc.pl.violin(adata=celegans_data,
keys=['Fst'],
groupby='embryo.time.bin.cat',
rotation=90,
palette='Paired',
stripplot=False,
inner='box',
order=['< 100', '100-130', '130-170', '170-210',
'210-270', '270-330', '330-390', '450-510',
'510-580', '580-650', '> 650'],
show=False)
ax.set_title('C. elegans - Fst distribution per embryo time')
plt.show()
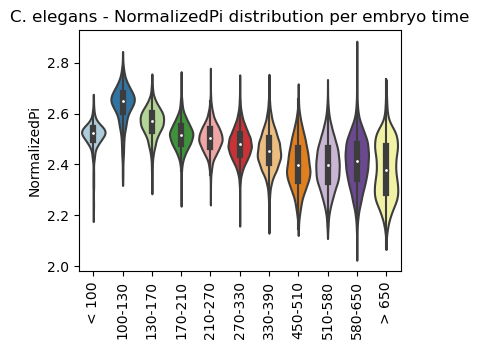
Boxplot NormalizedPi class per sample timepoint
[26]:
ax = sc.pl.violin(adata=celegans_data,
keys=['NormalizedPi'],
groupby='embryo.time.bin.cat',
rotation=90,
palette='Paired',
stripplot=False,
inner='box',
order=['< 100', '100-130', '130-170', '170-210',
'210-270', '270-330', '330-390', '450-510',
'510-580', '580-650', '> 650'],
show=False)
ax.set_title('C. elegans - NormalizedPi distribution per embryo time')
plt.show()
Boxplot gene age class per sample timepoint and add significance
Note: Please change notebook cell from raw to code to see the plots.
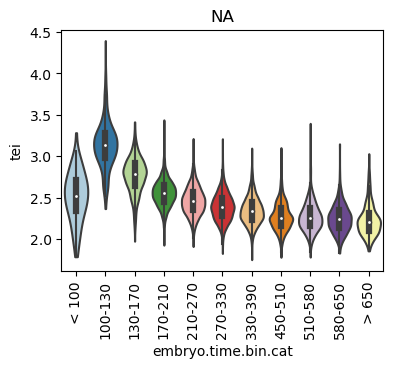
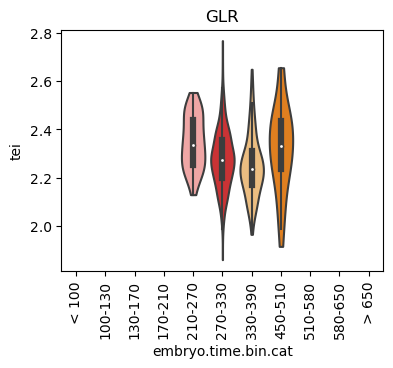
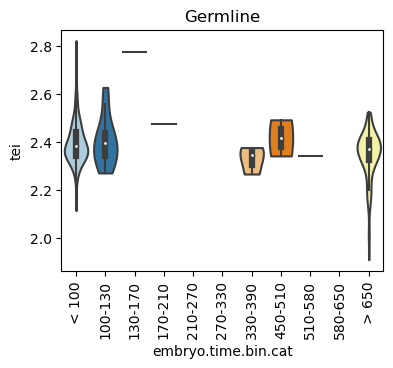
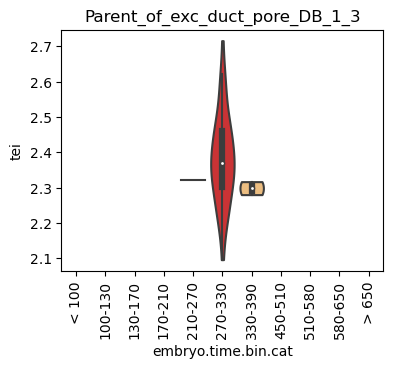
Boxplot gene age class per sample timepoint and per cell type
E.g. to just show the same plot for a selected cell-type, one could do the following.
List all annotated cell types:
[27]:
list(set(celegans_data.obs['cell.type']))
[27]:
['Excretory_duct_and_pore',
'Parent_of_exc_gland_AVK',
'Pharyngeal_intestinal_valve',
'M_cell',
'Pharyngeal_muscle',
'hmc',
'T',
'Coelomocyte',
'Z1_Z4',
'Seam_cell',
'G2_and_W_blasts',
'Body_wall_muscle',
'Hypodermis',
'Pharyngeal_neuron',
'Pharyngeal_gland',
'hmc_homolog',
'Intestinal_and_rectal_muscle',
'Ciliated_non_amphid_neuron',
'Excretory_cell_parent',
'ABarpaaa_lineage',
'Arcade_cell',
'Excretory_gland',
'XXX',
'Pharyngeal_marginal_cell',
'Excretory_cell',
'hmc_and_homolog',
'NA',
'GLR',
'Germline',
'Parent_of_exc_duct_pore_DB_1_3',
'hyp1V_and_ant_arc_V',
'Glia',
'Intestine',
'Ciliated_amphid_neuron',
'Rectal_cell',
'Rectal_gland',
'Parent_of_hyp1V_and_ant_arc_V']
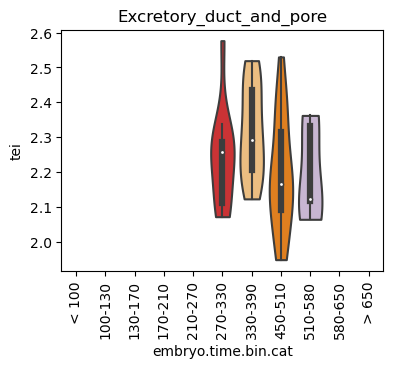
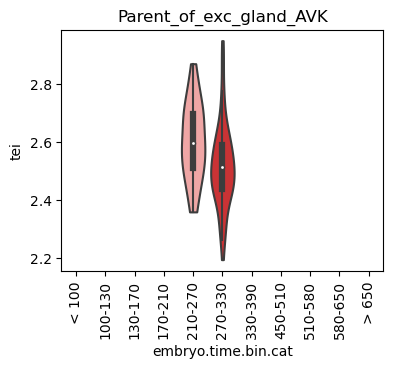
Loop over all cell types:
Note: Please change notebook cell from raw to code to see the plots.
[28]:
for ct in list(set(celegans_data.obs['cell.type'])):
ax = sc.pl.violin(adata=celegans_data[celegans_data.obs['cell.type']==ct],
keys=['tei'],
groupby='embryo.time.bin.cat',
rotation=90,
palette='Paired',
stripplot=False,
inner='box',
order=['< 100', '100-130', '130-170', '170-210',
'210-270', '270-330', '330-390', '450-510',
'510-580', '580-650', '> 650'],
show=False)
ax.set_title(ct)
ax.set_xlabel('embryo.time.bin.cat')
plt.show()
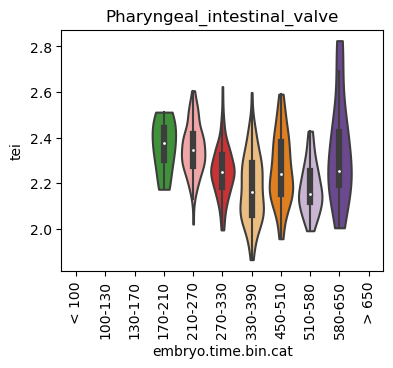
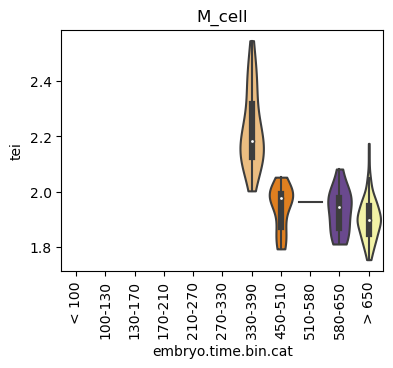
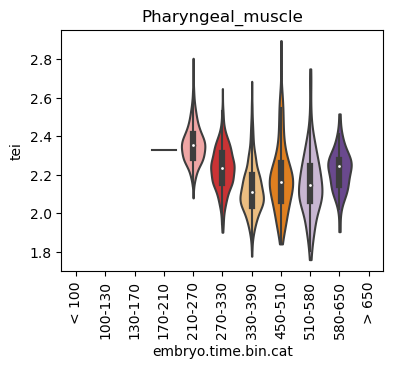
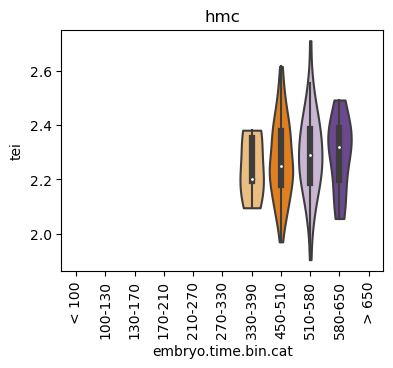
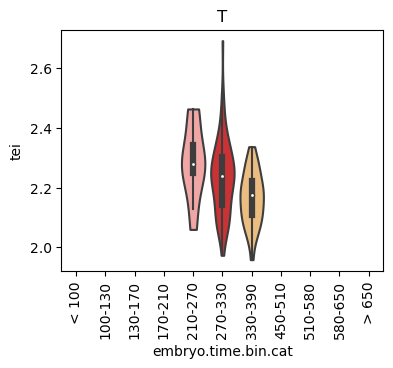
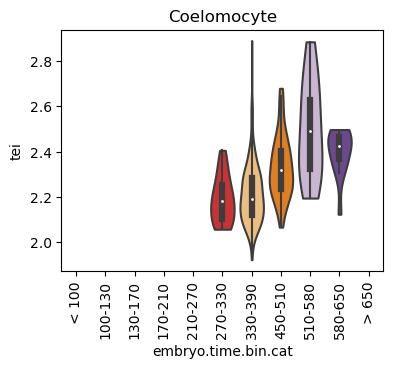
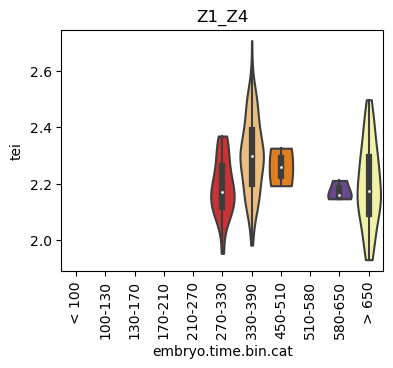
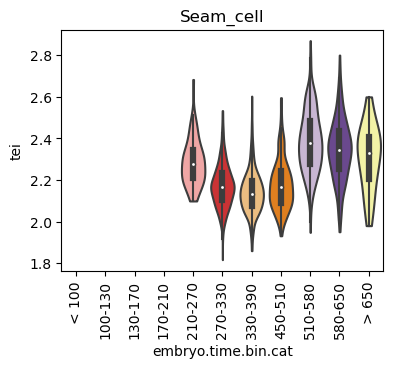
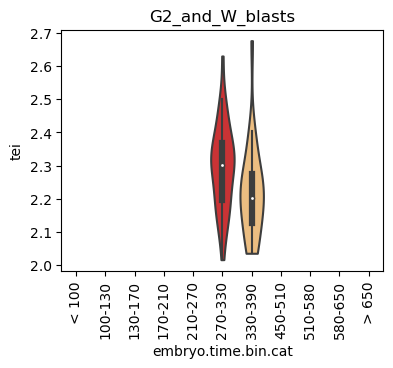
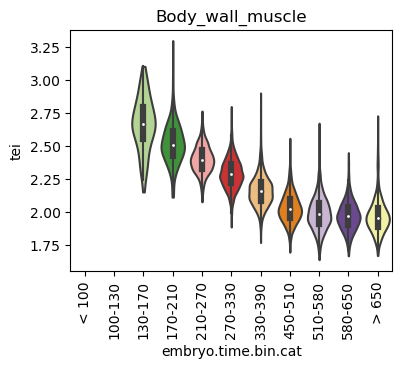
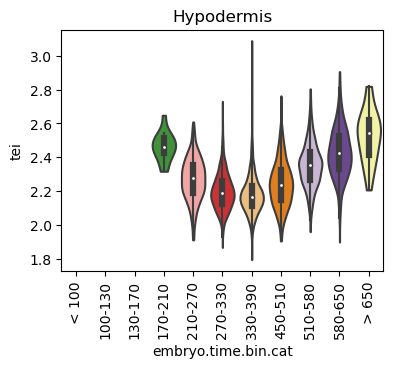
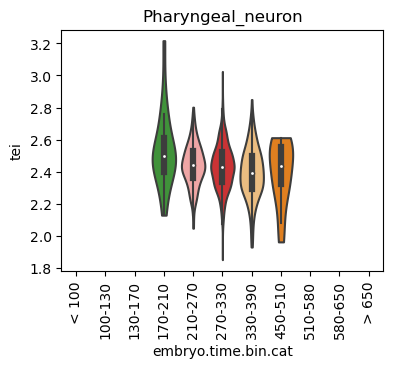
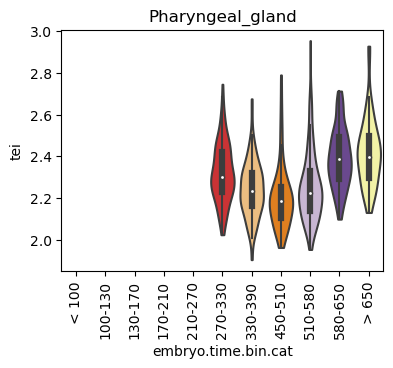
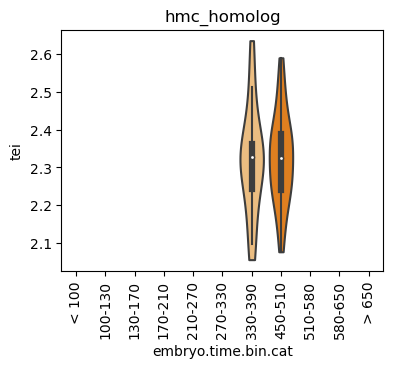
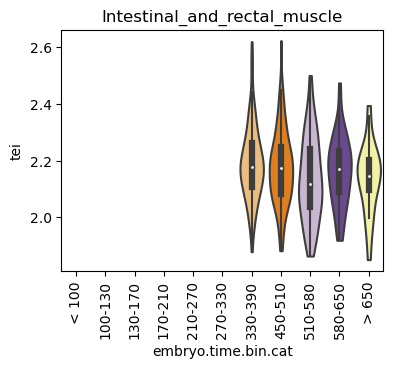
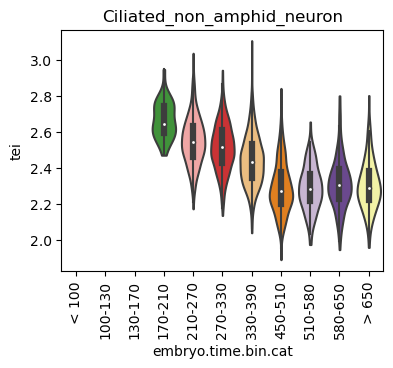
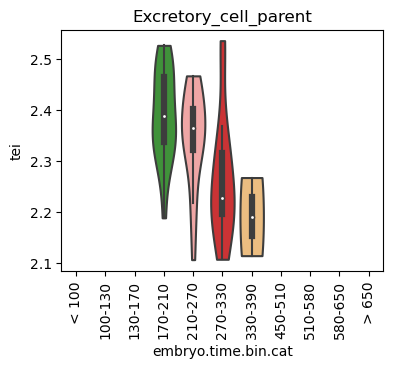
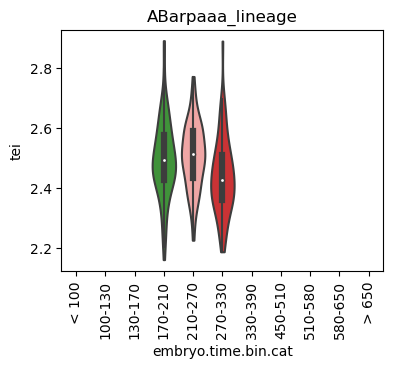
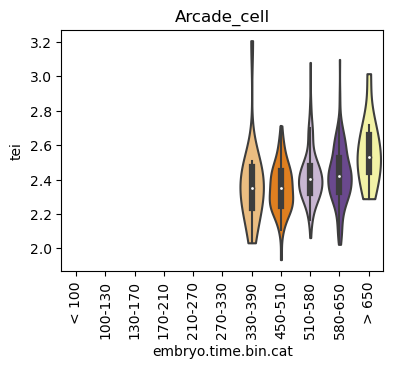
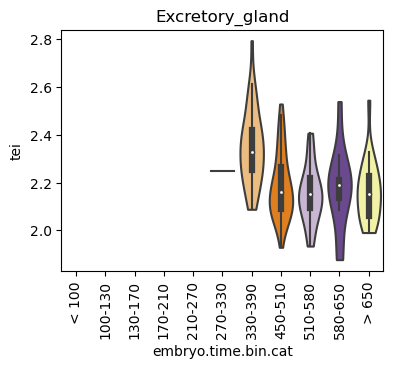
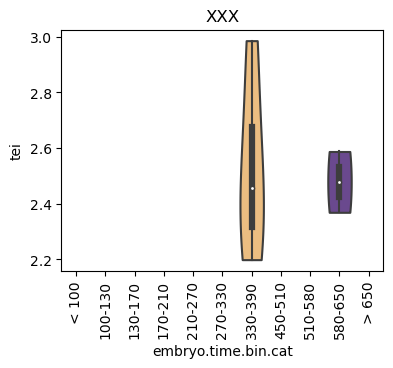
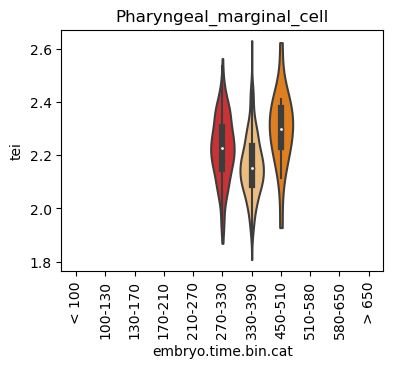
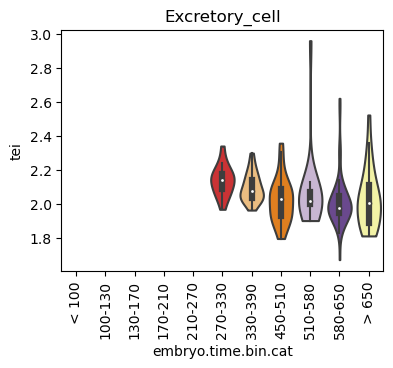
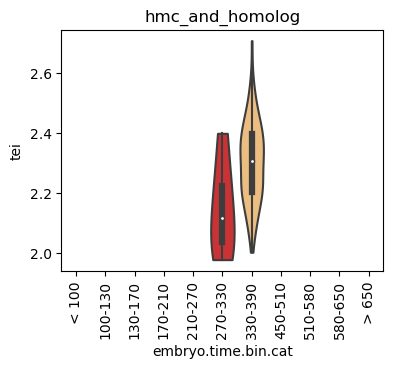
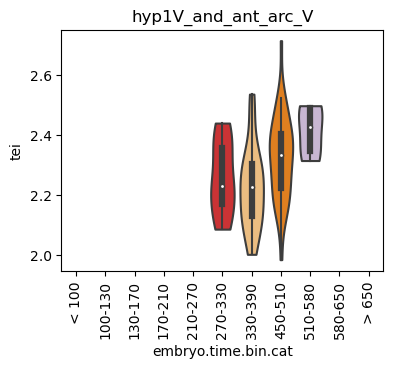
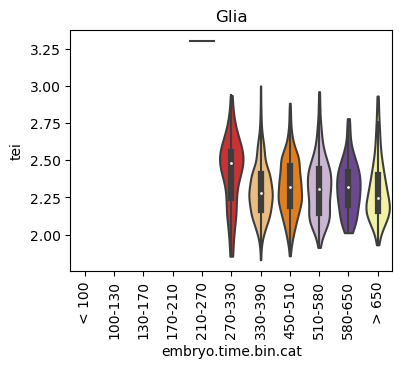
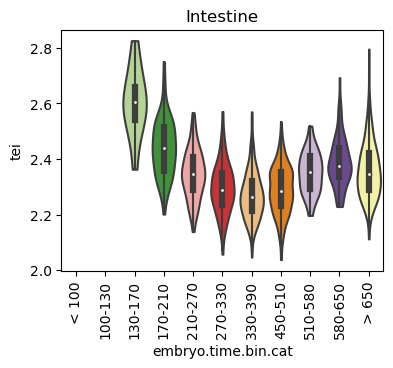
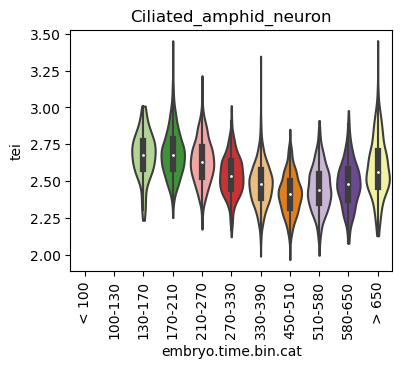
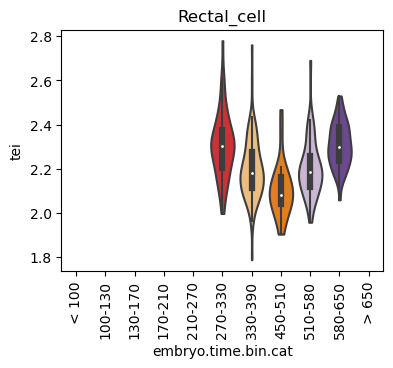
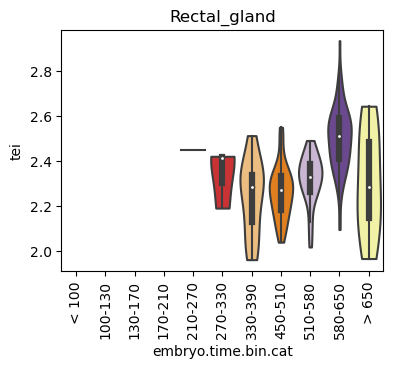
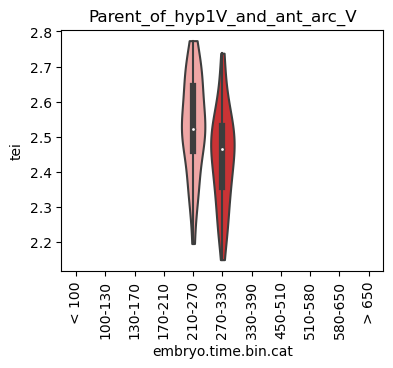
Scatterplot TEI vs TajimaD and color each cell by sample timepoint per cell type
Note: Please change notebook cell from raw to code to see the plots.
Scatterplot TEI vs Fst and color each cell by sample timepoint per cell type
Note: Please change notebook cell from raw to code to see the plots.
Scatterplot TEI vs NormalizedPi and color each cell by sample timepoint per cell type
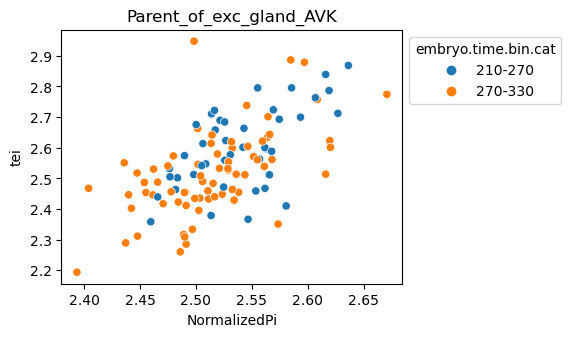
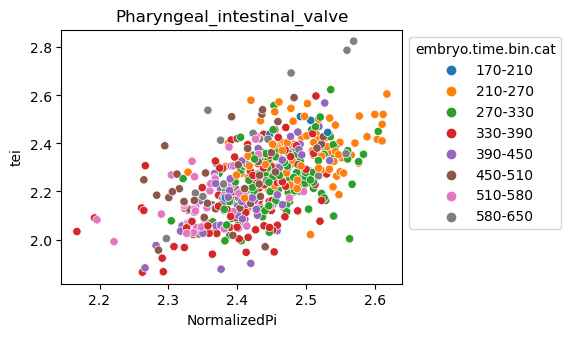
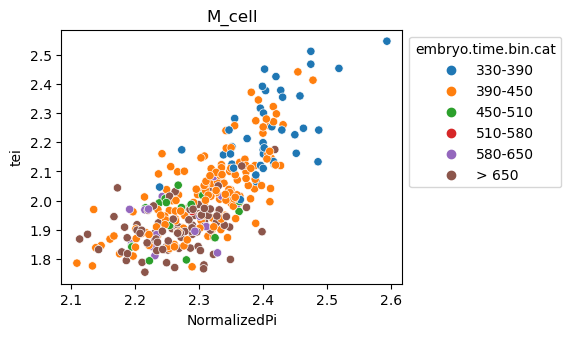
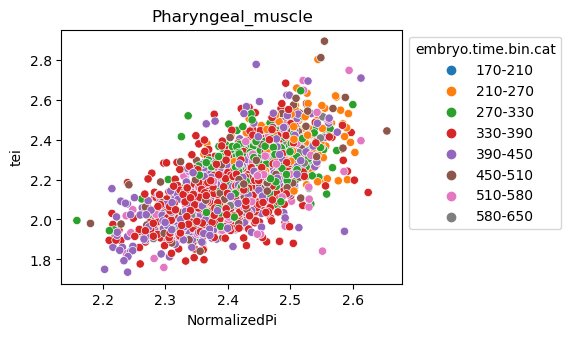
Note: Please change notebook cell from raw to code to see the plots.
[29]:
for ct in list(set(celegans_data.obs['cell.type'])):
ax = sns.scatterplot(data=celegans_data[celegans_data.obs['cell.type']==ct].obs,
x='NormalizedPi',
y='tei',
hue='embryo.time.bin.cat')
ax.set_title(ct)
sns.move_legend(ax, 'upper left', bbox_to_anchor=(1, 1))
plt.show()
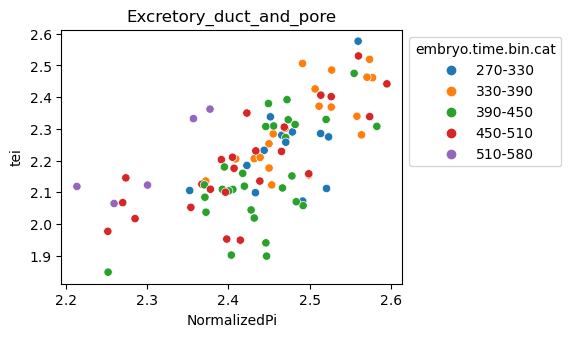
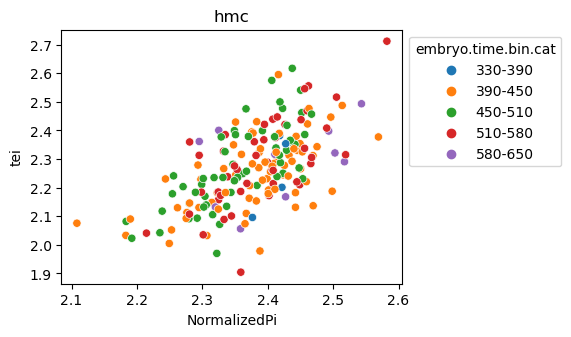
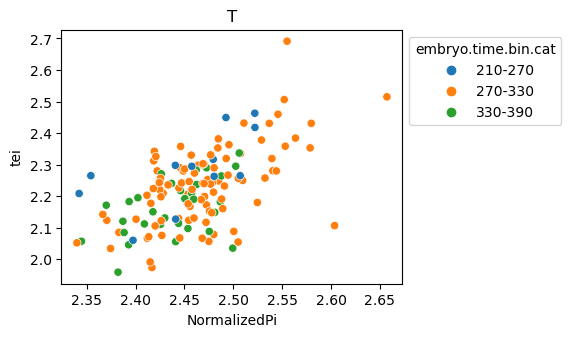
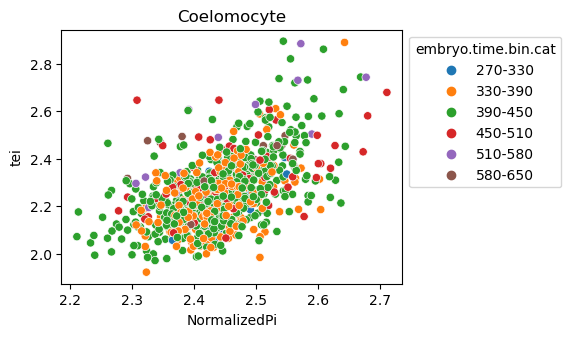
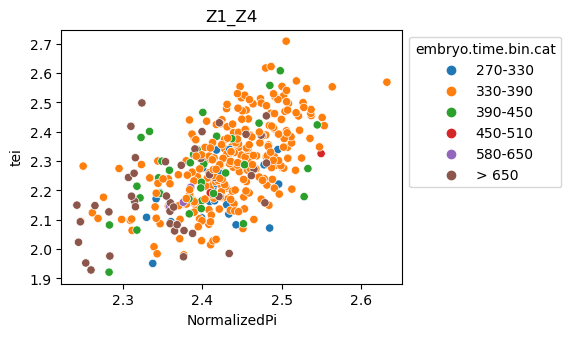
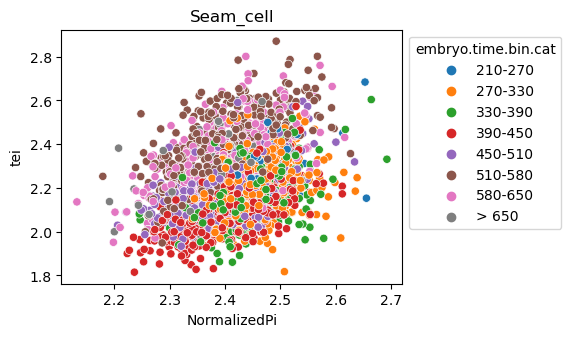
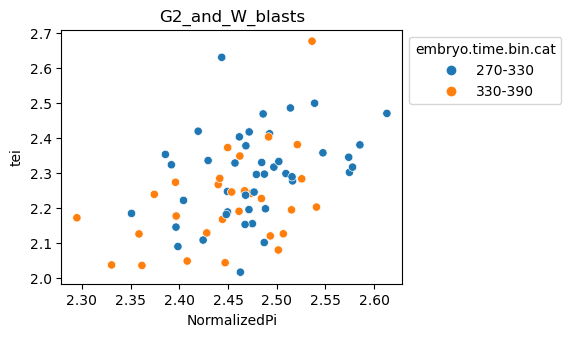
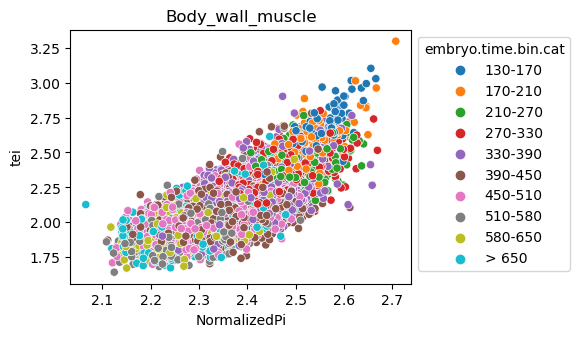
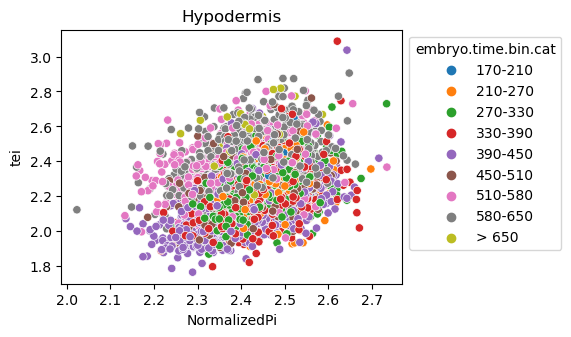
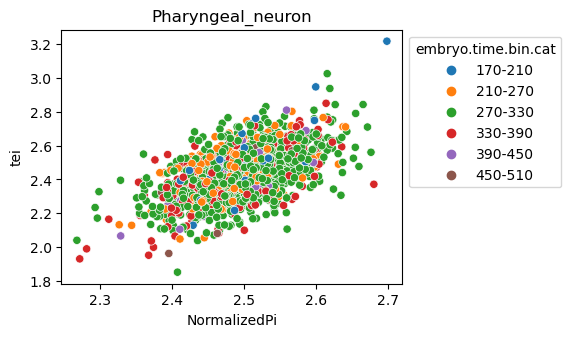
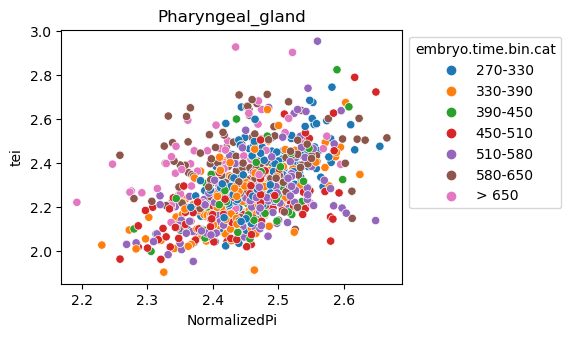
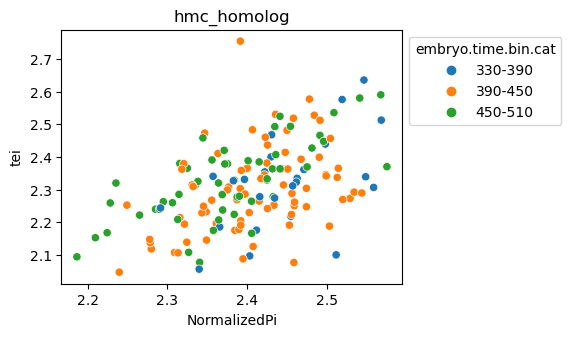
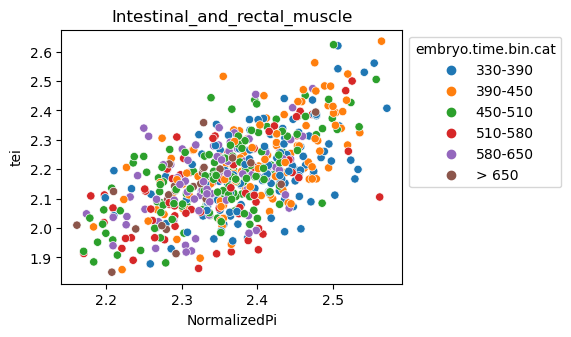
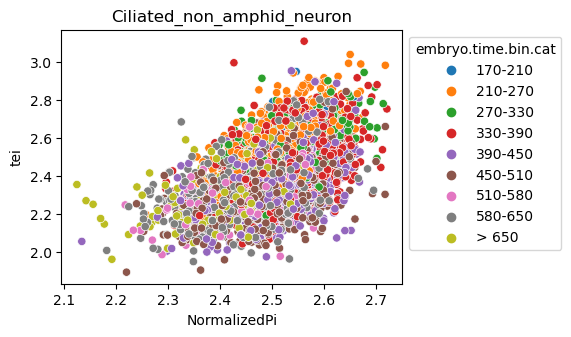
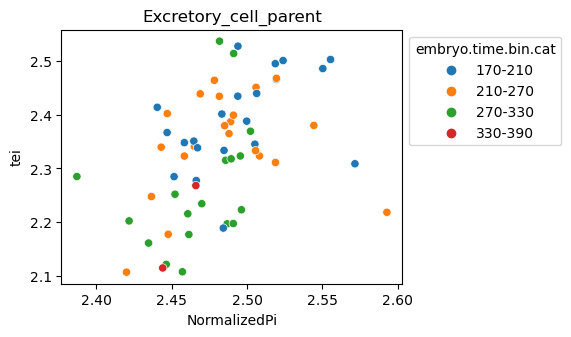
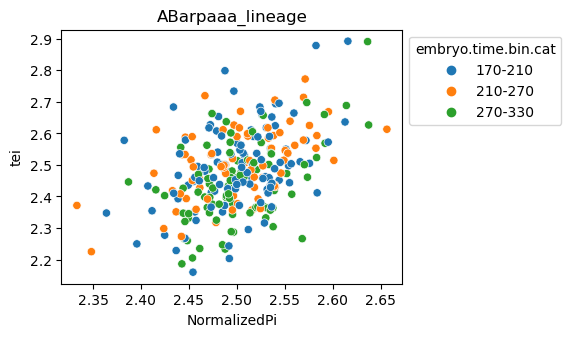
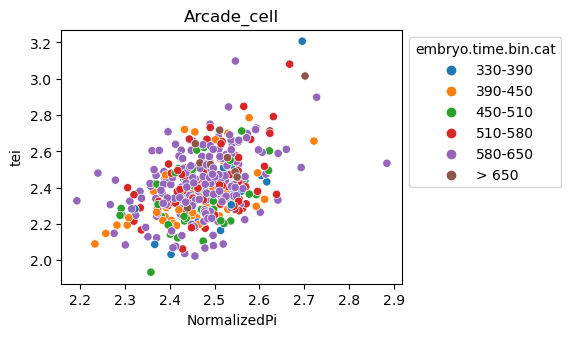
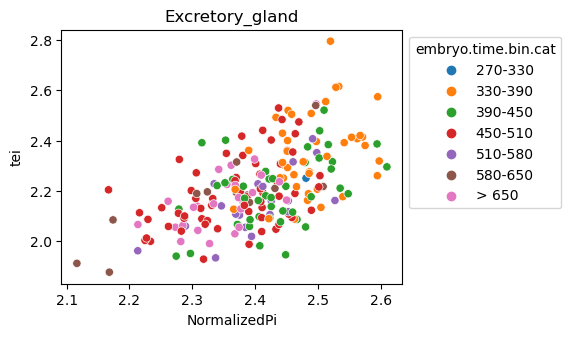
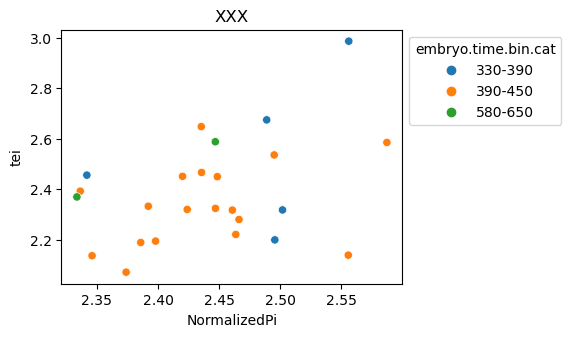
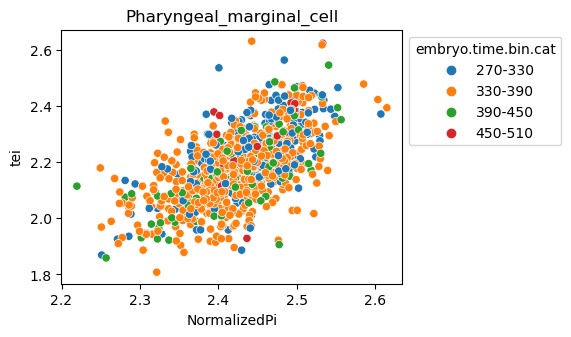
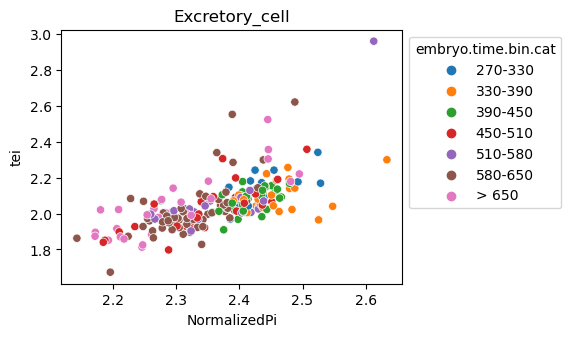
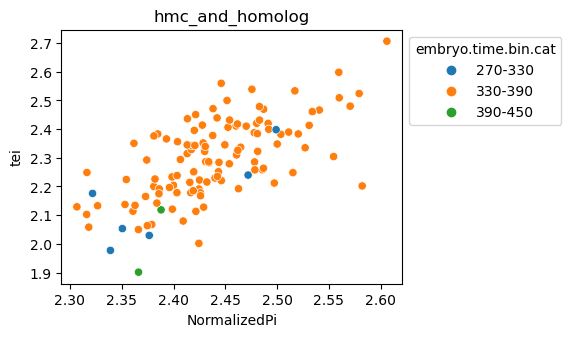
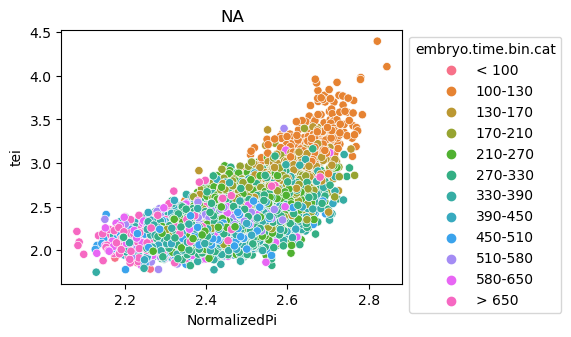
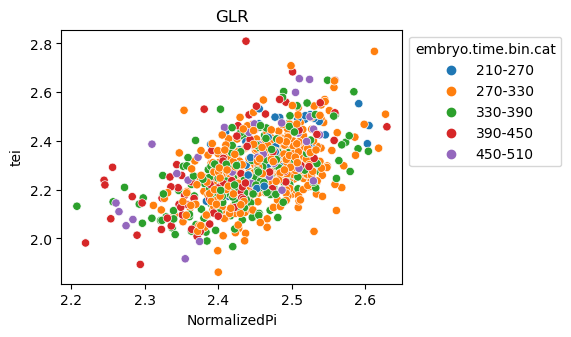
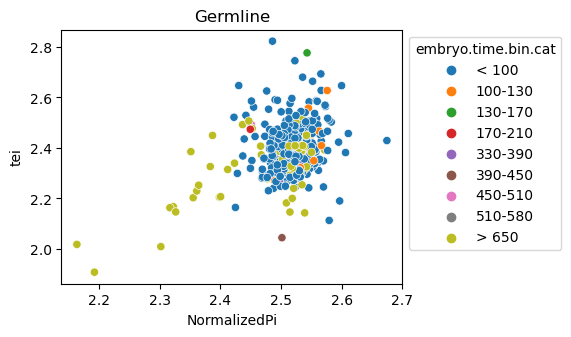
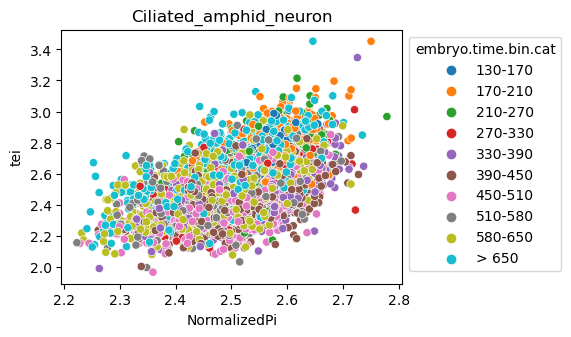
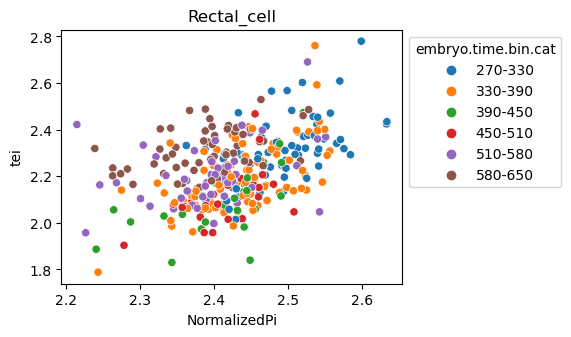
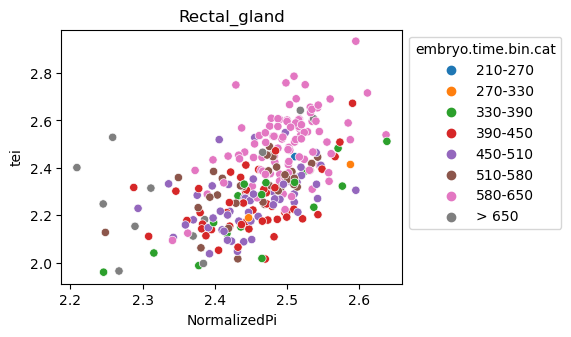
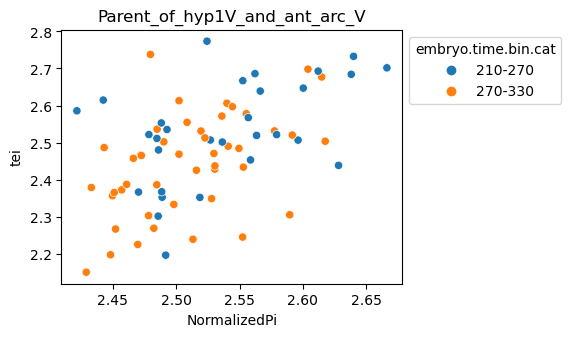
Plot relative expression per gene age class per sample timepoint
[30]:
celegans_data_rematrix_grouped = orthomap2tei.get_rematrix(
adata=celegans_data,
gene_id=query_orthomap['GeneID'],
gene_age=query_orthomap['Phylostratum'],
keep='min',
layer=None,
use='counts',
var_type='mean',
group_by_obs='embryo.time.bin.cat',
obs_fillna='__NaN',
obs_type='mean',
standard_scale=0,
normalize_total=True,
log1p=True,
target_sum=1e6)
celegans_data_rematrix_grouped
[30]:
| embryo.time.bin.cat | < 100 | 100-130 | 130-170 | 170-210 | 210-270 | 270-330 | 330-390 | 390-450 | 450-510 | 510-580 | 580-650 | > 650 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ps | ||||||||||||
| 0 | 0.717118 | 1.000000 | 0.584168 | 0.310426 | 0.302204 | 0.260612 | 0.206768 | 0.105727 | 0.048908 | 0.000000 | 0.013732 | 0.073366 |
| 1 | 1.000000 | 0.547242 | 0.477675 | 0.295885 | 0.250502 | 0.231566 | 0.176154 | 0.112925 | 0.089850 | 0.033621 | 0.000000 | 0.165599 |
| 2 | 1.000000 | 0.450475 | 0.387854 | 0.213375 | 0.168202 | 0.154225 | 0.113436 | 0.062726 | 0.048594 | 0.026063 | 0.000000 | 0.171621 |
| 3 | 1.000000 | 0.456666 | 0.312041 | 0.126569 | 0.089371 | 0.076377 | 0.078763 | 0.018744 | 0.046053 | 0.002353 | 0.000000 | 0.394810 |
| 4 | 1.000000 | 0.351679 | 0.245304 | 0.080961 | 0.056258 | 0.050955 | 0.028812 | 0.000000 | 0.017548 | 0.057651 | 0.015902 | 0.256118 |
| 5 | 1.000000 | 0.454323 | 0.381592 | 0.113054 | 0.024290 | 0.000000 | 0.019198 | 0.011902 | 0.006195 | 0.088254 | 0.077768 | 0.503433 |
| 6 | 1.000000 | 0.393570 | 0.331241 | 0.082090 | 0.019724 | 0.015556 | 0.019509 | 0.000000 | 0.010253 | 0.100935 | 0.250751 | 0.605489 |
| 7 | 1.000000 | 0.404537 | 0.296849 | 0.164328 | 0.114308 | 0.089470 | 0.035970 | 0.005399 | 0.000000 | 0.015025 | 0.024354 | 0.153953 |
| 8 | 1.000000 | 0.374614 | 0.332113 | 0.229286 | 0.175440 | 0.116940 | 0.052691 | 0.001487 | 0.004222 | 0.003206 | 0.000000 | 0.105183 |
| 9 | 1.000000 | 0.421409 | 0.314549 | 0.205528 | 0.172487 | 0.141735 | 0.098434 | 0.040396 | 0.017756 | 0.011292 | 0.000000 | 0.105804 |
| 10 | 1.000000 | 0.473084 | 0.224513 | 0.014137 | 0.000000 | 0.042069 | 0.067210 | 0.077679 | 0.173604 | 0.316754 | 0.270774 | 0.566895 |
| 11 | 0.613516 | 1.000000 | 0.562404 | 0.273833 | 0.202082 | 0.145245 | 0.092380 | 0.043486 | 0.033280 | 0.000000 | 0.003208 | 0.043322 |
| 12 | 0.470709 | 1.000000 | 0.665627 | 0.351440 | 0.257487 | 0.176988 | 0.101492 | 0.025500 | 0.011935 | 0.000000 | 0.000159 | 0.055289 |
| 13 | 0.411756 | 1.000000 | 0.649935 | 0.344974 | 0.252738 | 0.172185 | 0.096022 | 0.034795 | 0.025026 | 0.004728 | 0.000000 | 0.057878 |
[31]:
ax = sns.lineplot(celegans_data_rematrix_grouped.transpose(), palette='tab20', dashes=False)
ax.legend(fontsize=5, title='age class')
ax.set_title('C. elegans - Relative expression per embryo time')
ax.set_xlabel('embryo.time.bin')
ax.set_ylabel('Relative expression level')
sns.move_legend(ax, 'upper left', bbox_to_anchor=(1, 1))
plt.xticks(rotation=90)
plt.show()
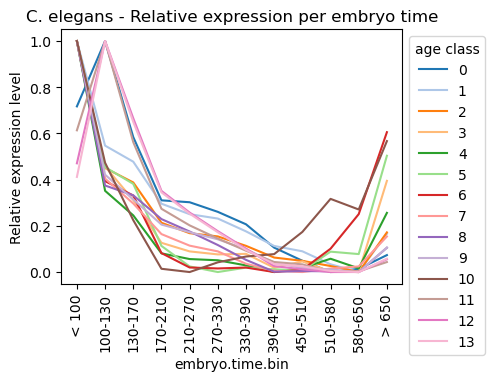
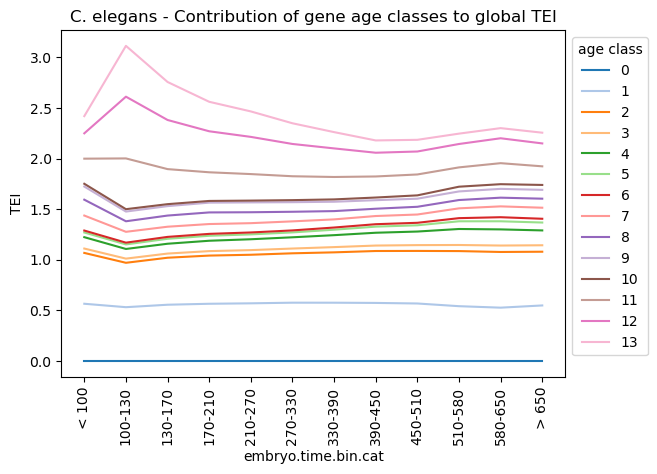
Get partial TEI values to visualize gene age class contributions
Partial TEI values can give an idea about which gene age class contributed at most to the global TEI pattern.
In detail, each gene gets a TEI contribution profile as follows:
\({TEI_{is} = f_{is} * ps_i}\)
, where \({TEI_{is}}\) is the partial TEI value of gene \({i}\), \({f_{is} = e_{is} / \sum e_{is}}\) and \({ps_i}\) is the phylostratum of gene i.
\({TEI_{is}}\) values are combined per \({ps}\).
The partial TEI values combined per strata give an overall impression of the contribution of each strata to the global TEI pattern.
One can either start from counts (adata.X) which is set as default or any other layer defined by the layer option (layer=None).
In addition, the counts can be normalized and log-transformed prior calculating partial TEI values (normalize_total=False, log1p=False, target_sum=1e6).
Further, these values can be combined per given observation, e.g. sample timepoint (group_by='raw.embryo.time').
The get_pstrata function of the orthomap2tei submodule will return two matrix, the first contains the sum of each partial TEI per gene age class and the second the corresponding frequencies.
Both can be further processed by returning the cumsum over the gene age classes. To get them set the option cumsum=True. The cumsum will result in either for the first matrix the TEI value per cell or mean TEI value per group, if one choose a observation with the group_by option. Or in case of the second frequency matrix will result in 1.
With the standard_scale option either gene age classes (standard_scale=0 rows) or cells or groups (standard_scale=1 columns) can be scaled, subtract the minimum and divide each by its maximum. By default no scaling is applied (standard_scale=None).
The resulting data will be visualized in the downstream section.
[32]:
celegans_pstrata = orthomap2tei.get_pstrata(adata=celegans_data,
gene_id=query_orthomap['GeneID'],
gene_age=query_orthomap['Phylostratum'],
keep='min',
layer=None,
cumsum=True,
group_by_obs='embryo.time.bin.cat',
obs_fillna='__NaN',
obs_type='mean',
standard_scale=None,
normalize_total=True,
log1p=True,
target_sum=1e6)
celegans_pstrata[0]
[32]:
| embryo.time.bin.cat | < 100 | 100-130 | 130-170 | 170-210 | 210-270 | 270-330 | 330-390 | 390-450 | 450-510 | 510-580 | 580-650 | > 650 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ps | ||||||||||||
| 0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
| 1 | 0.564379 | 0.530493 | 0.554769 | 0.564274 | 0.568691 | 0.574967 | 0.574761 | 0.572603 | 0.567071 | 0.540339 | 0.525781 | 0.547670 |
| 2 | 1.066948 | 0.969366 | 1.019430 | 1.040665 | 1.048817 | 1.063191 | 1.072649 | 1.085847 | 1.086674 | 1.085105 | 1.076311 | 1.078825 |
| 3 | 1.110209 | 1.010589 | 1.060572 | 1.084848 | 1.094679 | 1.110265 | 1.124388 | 1.139774 | 1.144051 | 1.145294 | 1.139959 | 1.142614 |
| 4 | 1.222601 | 1.106044 | 1.157717 | 1.187257 | 1.202083 | 1.221465 | 1.242487 | 1.266474 | 1.278308 | 1.303513 | 1.299550 | 1.289389 |
| 5 | 1.269068 | 1.150924 | 1.206516 | 1.236391 | 1.250166 | 1.269788 | 1.296496 | 1.326829 | 1.340107 | 1.379603 | 1.379764 | 1.367366 |
| 6 | 1.288148 | 1.168326 | 1.225392 | 1.255229 | 1.269197 | 1.289450 | 1.318017 | 1.350846 | 1.364751 | 1.410713 | 1.419942 | 1.404574 |
| 7 | 1.436406 | 1.275586 | 1.325999 | 1.352471 | 1.361118 | 1.378901 | 1.398513 | 1.431710 | 1.446642 | 1.507610 | 1.527176 | 1.513817 |
| 8 | 1.593160 | 1.379820 | 1.436263 | 1.467291 | 1.469098 | 1.473799 | 1.480398 | 1.503826 | 1.523043 | 1.589848 | 1.613239 | 1.603044 |
| 9 | 1.723074 | 1.475801 | 1.529523 | 1.564003 | 1.566499 | 1.568462 | 1.572929 | 1.588345 | 1.603053 | 1.674875 | 1.700259 | 1.690460 |
| 10 | 1.750491 | 1.499340 | 1.549016 | 1.580853 | 1.584175 | 1.588804 | 1.596092 | 1.614562 | 1.635971 | 1.721640 | 1.746472 | 1.738911 |
| 11 | 1.998728 | 2.001177 | 1.895514 | 1.864111 | 1.846383 | 1.824879 | 1.817629 | 1.822785 | 1.842534 | 1.912595 | 1.954244 | 1.922988 |
| 12 | 2.249283 | 2.611671 | 2.381169 | 2.269536 | 2.214056 | 2.144069 | 2.100194 | 2.057414 | 2.069561 | 2.143782 | 2.200446 | 2.149454 |
| 13 | 2.419276 | 3.114069 | 2.756838 | 2.561030 | 2.465832 | 2.348615 | 2.261164 | 2.178997 | 2.185036 | 2.246225 | 2.300599 | 2.255455 |
corresponds to main figure Figure 1E
[33]:
plt.rcParams['figure.figsize'] = [6.5, 4.5]
ax = sns.lineplot(celegans_pstrata[0].transpose(), palette='tab20', dashes=False)
ax.legend(fontsize=3, title='age class')
ax.set_title('C. elegans - Contribution of gene age classes to global TEI')
ax.set_xlabel('embryo.time.bin.cat')
ax.set_ylabel('TEI')
sns.move_legend(ax, 'upper left', bbox_to_anchor=(1, 1))
plt.xticks(rotation=90)
plt.show()
plt.rcParams['figure.figsize'] = [4.4, 3.3]
partial TajimaD
Note: Please change notebook cell from raw to code to see the plots.
Note: Please change notebook cell from raw to code to see the plots.
partial Fst
Note: Please change notebook cell from raw to code to see the plots.
Note: Please change notebook cell from raw to code to see the plots.
partial NormalizedPi
Note: Please change notebook cell from raw to code to see the plots.
Note: Please change notebook cell from raw to code to see the plots.
Color UMAP/TSNE by TEI
Follwoing the basic tutorial of the Scanpy python toolkit (Wolf et al., 2018), one can highlight TEI values on a dimensional reduction of the scRNA dataset, like PCA, UMAP or TSNE.
Filtering
[34]:
sc.pp.filter_genes(celegans_data, min_cells=3)
sc.pp.filter_cells(celegans_data, min_genes=200)
Normalization, Log transformation and Scaling
[35]:
sc.pp.normalize_total(celegans_data, target_sum=1e6)
sc.pp.log1p(celegans_data)
sc.pp.scale(celegans_data, max_value=10)
PCA and Neighbor calculations
[36]:
sc.tl.pca(celegans_data, svd_solver='arpack')
sc.pl.pca(celegans_data, color=['embryo.time.bin.cat', 'tei'])
/opt/anaconda3/envs/scanpy/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
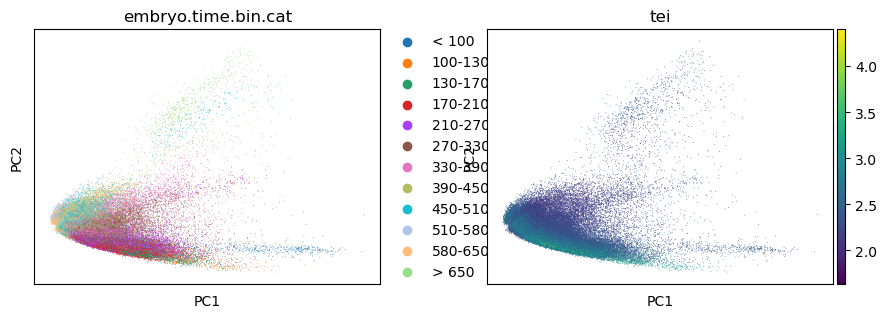
[37]:
sc.pp.neighbors(celegans_data)
Embedding the neighborhood graph
[38]:
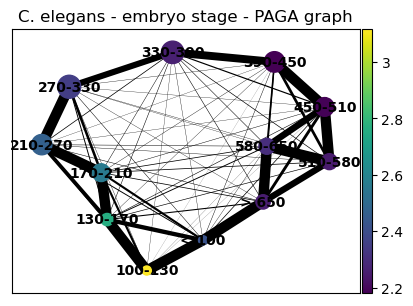
sc.tl.paga(celegans_data, groups='embryo.time.bin.cat')
sc.pl.paga(celegans_data, title='C. elegans - embryo stage - PAGA graph')
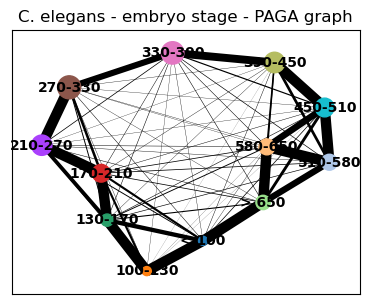
[39]:
sc.pl.paga(celegans_data, title='C. elegans - embryo stage - PAGA graph', color=['tei'])
UMAP
[40]:
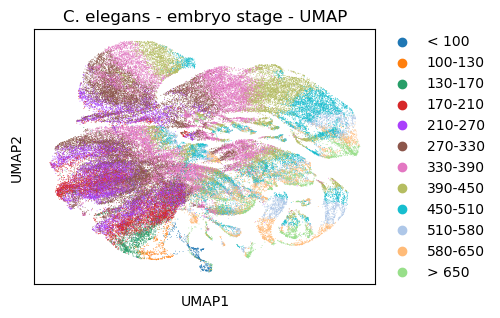
sc.tl.umap(celegans_data,
init_pos='paga')
sc.pl.umap(celegans_data,
title='C. elegans - embryo stage - UMAP', color=['embryo.time.bin.cat'])
/opt/anaconda3/envs/scanpy/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
Color UMAP by cell type
[41]:
sc.pl.umap(celegans_data,
title='C. elegans - cell type - UMAP', color=['cell.type'])
/opt/anaconda3/envs/scanpy/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(

Color UMAP by TEI
[42]:
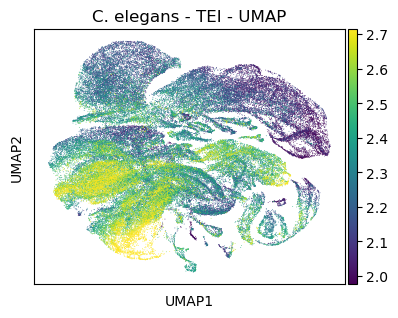
#plt.rcParams['figure.figsize'] = [7.5, 4.5]
sc.pl.umap(celegans_data,
title='C. elegans - TEI - UMAP',
color=['tei'],
color_map='viridis',
vmin='p5',
vmax='p95')
#plt.rcParams['figure.figsize'] = [6, 4.5]
Color UMAP by TajimaD bins
[43]:
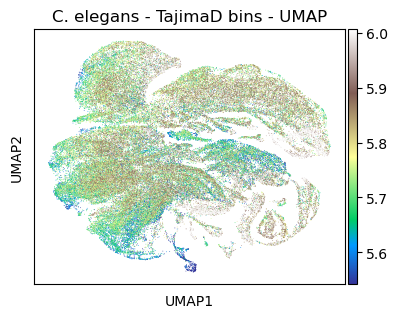
#plt.rcParams['figure.figsize'] = [7.5, 4.5]
sc.pl.umap(celegans_data,
title='C. elegans - TajimaD bins - UMAP',
color=['TajimaD'],
color_map='terrain',
vmin='p5',
vmax='p95')
#plt.rcParams['figure.figsize'] = [6, 4.5]
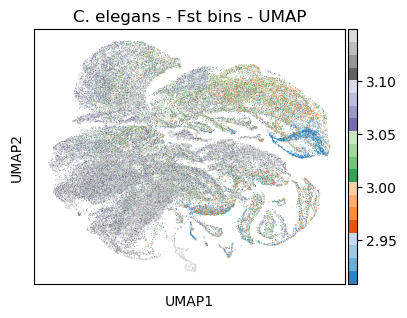
Color UMAP by Fst bins
[44]:
#plt.rcParams['figure.figsize'] = [7.5, 4.5]
sc.pl.umap(celegans_data,
title='C. elegans - Fst bins - UMAP',
color=['Fst'],
color_map='tab20c',
vmin='p5',
vmax='p95')
#plt.rcParams['figure.figsize'] = [6, 4.5]
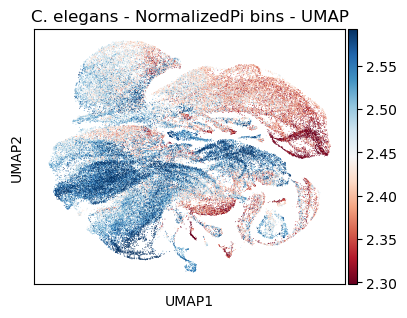
Color UMAP by NormalizedPi bins
[45]:
#plt.rcParams['figure.figsize'] = [7.5, 4.5]
sc.pl.umap(celegans_data,
title='C. elegans - NormalizedPi bins - UMAP',
color=['NormalizedPi'],
color_map='RdBu',
vmin='p5',
vmax='p95')
#plt.rcParams['figure.figsize'] = [6, 4.5]
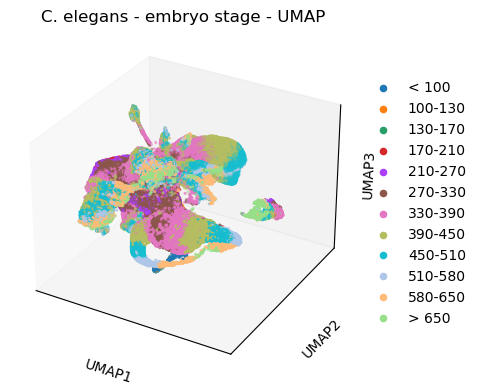
3D-UMAP
[46]:
plt.rcParams['figure.figsize'] = [7.5, 4.5]
#3d
sc.tl.umap(celegans_data,
n_components=3)
sc.pl.umap(celegans_data,
title='C. elegans - embryo stage - UMAP',
color=['embryo.time.bin.cat'],
projection='3d')
plt.rcParams['figure.figsize'] = [4.4, 3.3]
/opt/anaconda3/envs/scanpy/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:325: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = ax.scatter(
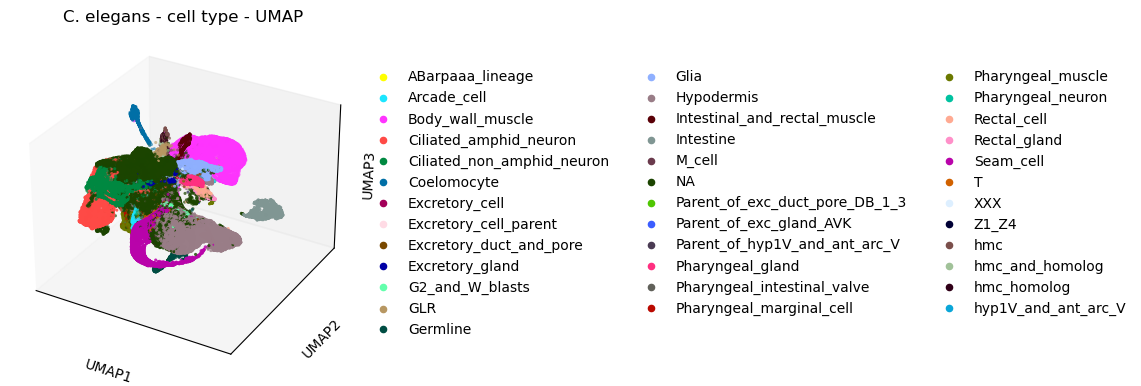
[47]:
plt.rcParams['figure.figsize'] = [7.5, 4.5]
#3d
sc.pl.umap(celegans_data,
title='C. elegans - cell type - UMAP',
color=['cell.type'],
projection='3d')
plt.rcParams['figure.figsize'] = [4.4, 3.3]
/opt/anaconda3/envs/scanpy/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:325: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = ax.scatter(
[48]:
plt.rcParams['figure.figsize'] = [7.5, 4.5]
#3d
sc.pl.umap(celegans_data,
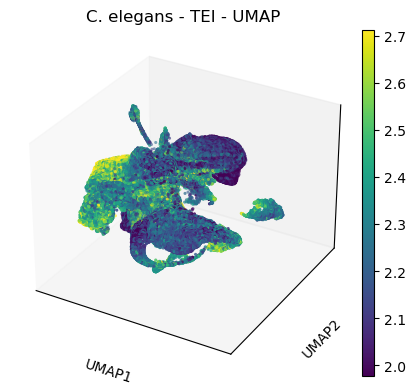
title='C. elegans - TEI - UMAP',
color=['tei'],
color_map='viridis',
vmin='p5',
vmax='p95',
projection='3d')
plt.rcParams['figure.figsize'] = [4.4, 3.3]
[49]:
plt.rcParams['figure.figsize'] = [7.5, 4.5]
#3d
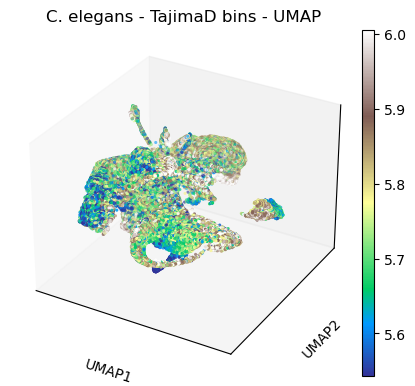
sc.pl.umap(celegans_data,
title='C. elegans - TajimaD bins - UMAP',
color=['TajimaD'],
color_map='terrain',
vmin='p5',
vmax='p95',
projection='3d')
plt.rcParams['figure.figsize'] = [4.4, 3.3]
[50]:
plt.rcParams['figure.figsize'] = [7.5, 4.5]
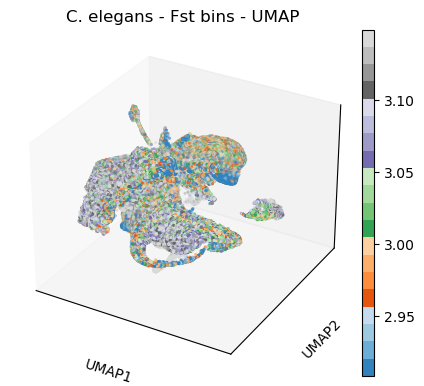
sc.pl.umap(celegans_data,
title='C. elegans - Fst bins - UMAP',
color=['Fst'],
color_map='tab20c',
vmin='p5',
vmax='p95',
projection='3d')
plt.rcParams['figure.figsize'] = [4.4, 3.3]
[51]:
plt.rcParams['figure.figsize'] = [7.5, 4.5]
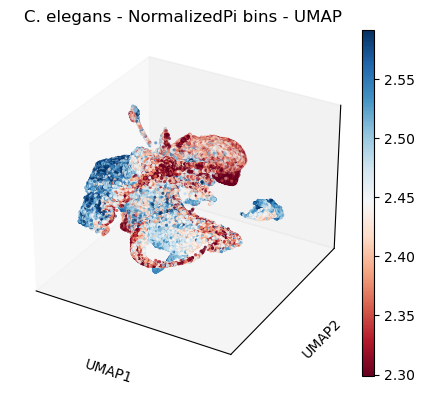
#3d
sc.pl.umap(celegans_data,
title='C. elegans - NormalizedPi bins - UMAP',
color=['NormalizedPi'],
color_map='RdBu',
vmin='p5',
vmax='p95',
projection='3d')
plt.rcParams['figure.figsize'] = [4.4, 3.3]
Please have a look at the documentation for other case studies.